Analysis Run Release Notes
- Former user (Deleted)
- Former user (Deleted)
- David Heiman
- Former user (Deleted)
Release notes for the standardized data upon which these analysis runs are based are available here
NOTE: The LIHC RPPA data submitted by MDACC early in 2016 were discovered to be mislabelled MESO samples. Thus the 2016_01_28 analyses and standard data pipelines for LIHC have been re-run using the corrected samples submitted in March, and the nozzle reports now contain notices of the discrepancies.
The following table shows the changes in sample counts for RPPA data as a result of this patch:
HNSC | +145 | (357 total) |
|---|---|---|
LIHC | +121 | (184 total) |
THCA | +146 | (368 total) |
This is likely to be either the penultimate or perhaps even final standard Firehose analysis run of the TCGA project. Custom AWG runs will continue for TCGA as needed.
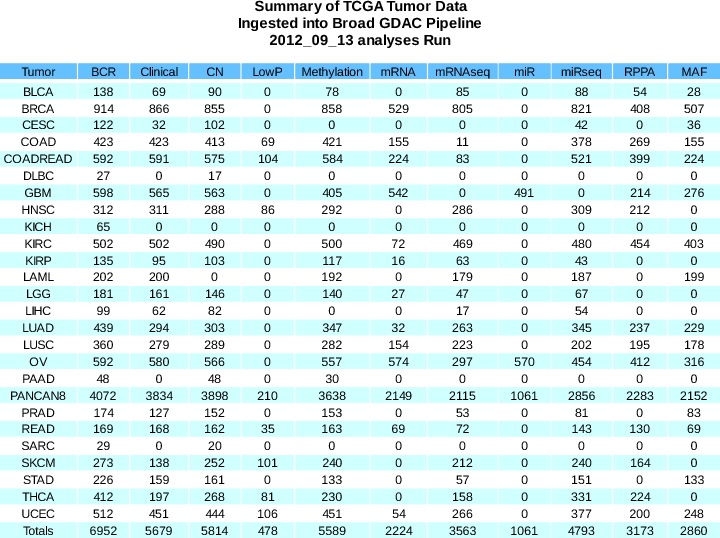
This analysis run was based upon the 2016_01_28 data run and includes 1528 analysis reports.
Summary of sample changes (see the comprehensive samples report for more details) since the Fall 2015 analysis run:
BCR
+1
(11368 total)
Clinical
+32
(11196 total)
CN
+2
(10987 total)
MAF
+313
(7099 total)
Methylation
+1
(10972 total)
miRSeq
+2
(10156 total)
mRNASeq
+164
(10267 total)
rawMAF
+2072
(6322 total)
RPPA
+627
(7429 total)
- APOBEC pipelines updated:
- used median filtering in primary APOBEC analysis
- in downstream clinical correlations, corrected names of categorical variables and descriptions of how they were utilized
- cNMF clustering improvement: new criteria used to select best cluster, identical to that describe in Summer 2014 run (see below) for consensus hierarchical clustering:
The cophenetic correlation coefficients and average silhouette values are used to determine the k with the most robust clusterings. From the plot of cophenetic correlation versus k, we select modes and the point preceding the greatest decrease in cophenetic correlation coefficient, and from these choose the k with the highest average silhouette value. - Survival analysis: for all clinical correlations
- Modified the p-value calculation of survival analysis with continuous data. It now uses the quantile interval categorical values instead of continuous values.
Previously it had one hazard ratio value for one continuous value, but now has multiple hazard ratio values for quantile interval curves (and are now reflected in the plot legends)
- FireBrowse:
- Updated to v1.1.28 to reflect these run results
- iCoMut:
- loaded 4 additional disease cohorts: DLBC, ESCA, SARC, and THYM
- Completed most of work for major new release, stay tuned for announcement next week, incorporating many graphical and data exploration enhancements
Summary of sample changes (see the comprehensive samples report for more details):
BCR +15 (11367 total) Clinical +219 (11164 total) CN -2 (10985 total) LowP +120 (1211 total) MAF +202 (6786 total) Methylation +16 (10971 total) miRSeq -6 (10154 total) mRNASeq +8 (10103 total) rawMAF +213 (4250 total) RPPA +1346 (6802 total) - Mutation Analyses:
- Stick figures now available in all MutSig reports due to addition of Oncotator to our standard data runs
- MutSig 1.5 removed. MutSig CV will be removed from our next analyses run
- coMut output tables will now be generated even if no mutation data exists for a given cohort;
- this enables iCoMut to still be used to quickly examine the rest of the analysis result profile
MutSig results for COAD/READ, LAML, and OV are not available for MutSig_2.0, the oldest MutSig algorithm in our Analyses run. They are available in both MutSig_CV and MutSig_2CV. This is due to our new policy of lifting over these MAFs from hg18 to hg19, which all other TCGA MAFs are currently based on, while we have not performed lliftovers on the accompanying coverage files, which MutSig_2.0 requires.
- APOBEC analysis now run on COAD/READ, OV, LAML due to new policy of lifting over to hg19
- Clinical analyses: names of some Clinical Data Elements (CDEs) have changed. See the Clinical Data section of our pipeline documentation for more details.
- FireBrowse 1.1.20:
- Loaded these 2015_08_21 analysis results
Small cleanup in analysis report accordion of UI: remove duplicate Pathway_Overlaps report, and shrink MutSig report name entries
Analyses/MAF api no longer shows results from MutSig_1.5
Analyses/FeatureTable and Analyses/Reports apis are now sorted in descending date order
Metadata/Platforms and Metadata/Centers apis are now sorted, too, for easier visual inspection
- iCoMut: many new features, including
- Significantly more advanced search: including logical inclusion/exclusion, and searching individual panel results using comparison operators (e.g. >, =, etc)
- Figure generation (download SVG), with embeddable link one can use to fully reproduce the figure
- Ability to insert additional genes into the mutation panel (as long as they are in the MutSig SMG list)
- Ability to display continuous data in addition to categorical (e.g. clinical age values are no longer just "hi/med/low" but actual numeric values)
- viewGene: several bug fixes and improvements to cross-browser support
Over 8000 new aliquots since the Fall 2014 analyses run: see the comprehensive samples report for more details
BCR
+38
(11352 total)
Clinical
+1352
(10945 total)
CN
+1
(10987 total)
MAF
+523
(6584 total)
Methylation
+533
(10955 total)
miRSeq
+109
(10160 total)
mRNASeq
+365
(10095 total)
rawMAF
+4250
(4250 total)
RPPA
+1120
(5456 total)
New rawMAF data type: for some disease studies, mutation samples continued to be collected and sequenced after the respective marker paper data freeze. Until now Firehose has packaged and run analyses only on the mutations in the data freeze for the submitted paper. However, since it's in the best interests of the community to have as many samples as possible for analysis, as of this run we are excited to announce that Firehose now includes MAFs that have accumulated post-publication; we introduce the term rawMAF to describe such samples, connoting that they have not necessarily been curated by a TCGA analysis working group. This effort has added over 1500 new samples to our data stream.
- Total of 1480 analysis result reports, an increase of 311 since the last analysis run.
- New analysis pipelines:
Pathway_GSEA_mRNAseq
Performs gene set enrichment analysis for mRNAseq clusters using Broad Institute GSEA MsigDB Class2 canonical pathways. Also inspects core enriched genes for each top significant gene set, and checks their expression fold-change level and significance by eBayes lm fit.Correlate_Clinical_vs_Mutation_APOBEC_Categorical
Checks correlation between clinical features and APOBEC groups classified into 3 sample groups of APOBEC high, low and none based on APOBEC MUTLOAD MINESTIMATE and APOBEC Enrichment score.Correlate_Clinical_vs_Mutation_APOBEC_Continuous
Checks correlation between APOBEC scores and selected clinical features.Correlate_mRNAseq_vs_Mutation_APOBEC
Attempts to calculate the pearson correlation between APOBEC_MutLoad_MinEstimate and mRnaseq data of each gene across samples.miRseq_FindDirectTargets
Infers putative direct gene targets of miRs based on miRseq and mRNAseq expression profiles across multiple samples.Mutation_CoOccurrence
Tailors the Firehose AnalysisFeatureTable specifically for the iCoMut visualization tool.Pathway_Overlaps_MSigDB_MutSig2CV
Checks pathway overlaps for significant genes identified by MutSig2CV using hypergeometric test.
- FireBrowse:
- iCoMut: a powerful new synoptic tool for interpretation and exploration CoMut figures have quickly become a staple of TCGA research. Within a single graphic they provide a comprehensive analysis profile, enabling the reader to quickly infer relationships between co-occurring results. With iCoMut researchers can now explore coMut figures interactively, sorting and reordering samples and results as they see fit.
- New viewGene expression visualizer: built on top of the FireBrowse RESTful API, viewGene generates a boxplot of mRNASeq expression levels for a selected gene across all cohorts.
- Reports and FeatureTable apis now make older results available, too, not merely those from the latest analysis run. The search criterion is YYYY_MM_DD datestamp.
- Many recent enhancements as described in April 2015 stddata run notes.
- Issues
- The new raw MAF COAD samples contain many InDels, which increased the number of SMGs identified for COAD[READ]: for signficance analysis we internally perform liftover of the COAD[READ] MAFs from hg18 to hg19. Typically this liftover has no appreciable impact on the SMGs identified in MAFs which largely contain simpler mutations, but for the InDel-heavy COAD[READ] MAFs the SMG lists have grown to upwards of 1000 genes. To alleviate this in future runs we will liftover and reannotate with the latest hg19 version of Gencode, using Oncotator in our standard data runs.
Mutation_APOBEC:
- Added input preprocessing to enable analysis to include X and Y chromosomes
- Cleaned up portions of the report and removed a redundant section
- Corrected archives to include all result files
Nearly 6000 aliquots added since the Summer 2014 analyses run (see the comprehensive samples report for more details):
Clinical +899 (9593 total) CN +1512 (10986 total) MAF +143 (6061 total) Methylation +513 (10422 total) miRSeq +1176 (10051 total) mRNASeq +1182 (9730 total) RPPA +303 (4336 total) - Number of analyses & reports increased from 1008 to 1164
- Including 52 analyses for 3 previously unanalyzed disease cohorts: CHOL, TGCT, and THYM (Cholangiocarcinoma, Testicular Germ Cell Tumors, Thymoma)
- New Analysis Pipelines:
Mutation_APOBEC : determines the level of APOBEC signature activity within mutations (Roberts et al 2013). Result reports are available for 21 disease cohorts, from either the FireBrowse UI or the Broad GDAC site. Selected portions of these results will also be programmatically accessible through the FireBrowse Analyses API in early 2015.
Aggregate_AnalysisFeatures: as described here, this pipeline aims to integrate within a single feature table the most important findings across the entire corpus of GDAC Firehose results. See the examples and code in these reports to learn how this can simplify downstream integrative analysis and ad-hoc exploration. The feature tables for each disease cohort may also be examined interactively in FireBrowse and queried programmatically through its API.
- Firebrowse api calls no longer require the :8000 port number to be present in the URL (although it is still accepted)
- Issue with RPPA Analyses for SKCM:
- The names in the "Composite Element REF" column of the antibody annotation file from the mdanderson.org_SKCM.MDA_RPPA_Core.mage-tab.1.5.0 archive do not match the same column for the RPPA files in the mdanderson.org_SKCM.MDA_RPPA_Core.Level_3.1.3.0 archive. This disabled gene name annotation, causing issues with clustering and correlations.
We are excited to announce that Firebrowse Beta 1 is available for public use. As described in the short tutorial, with this initial release our aim is to make it significantly easier to explore the large volume of datasets and analysis results produced by our TCGA GDAC Firehose, both interactively as well as programmatically through a set of over 20 nascent RESTful apis which support both coarse- and fine-grained queries. In the coming months we’ll nudge the API towards maturity while incorporating new analysis and visualization tools, so please visit often and don’t be shy about sharing your feedback!
Summary of sample changes since the Spring analyses run (see the comprehensive samples report for more details):
BCR
+1143
(11314 total)
Clinical
+604
(8694 total)
CN
+308
(9474 total)
MAF
+380
(5918 total)
Methylation
+681
(9909 total)
miRSeq
+674
(8875 total)
mRNASeq
+524
(8548 total)
RPPA
+1
(4033 total)
- Mutation Analyses:
- MutSig 2CV incorporated into Analyses workflow
- While older MutSig versions remain in Analysis workflow, MutSig2CV should be considered the preferred version
- As such, our Analyses workflow no longer merges the results of multiple MutSig versions into a single result for downstream integrative analyses (e.g. correlations)
- Instead, downstream integrative analyses utilize the MutSig2CV results (with one exception: correlation of mutation rate vs. clinical still uses Mutsig 2.0)
- Consensus hierarchical clustering improvements:
- Discontinued use of the much older Java implementation by Monti, et al, in favor of ConsensusClusterPlus R (v1.18.0) package by Wilkerson, et al
- New criteria used to select best cluster:
The cophenetic correlation coefficients and average silhouette values are used to determine the k with the most robust clusterings. From the plot of cophenetic correlation versus k, we select modes and the point preceding the greatest decrease in cophenetic correlation coefficient, and from these choose the k with the highest average silhouette value.
- GenePattern: we have migrated completely away from using GenePattern as a backend for running any tasks in GDAC Firehose
- Substantial improvements to clinical correlation pipelines:
- Race and ethnicity are now included in the list of selected tier1 clinical parameters for correlation analyses
- Nozzle reports now make clear that correlations against miR, miR-Seq, mRNA and mRNA-Seq data use log2 expression levels
- Discontinued use of parametric statistical tests in favor of non-parametric statistical tests:
T-test changed to Wilcoxon test, ANOVA changed to Kruskal-Wallis and Chi-square test to Fisher's exact test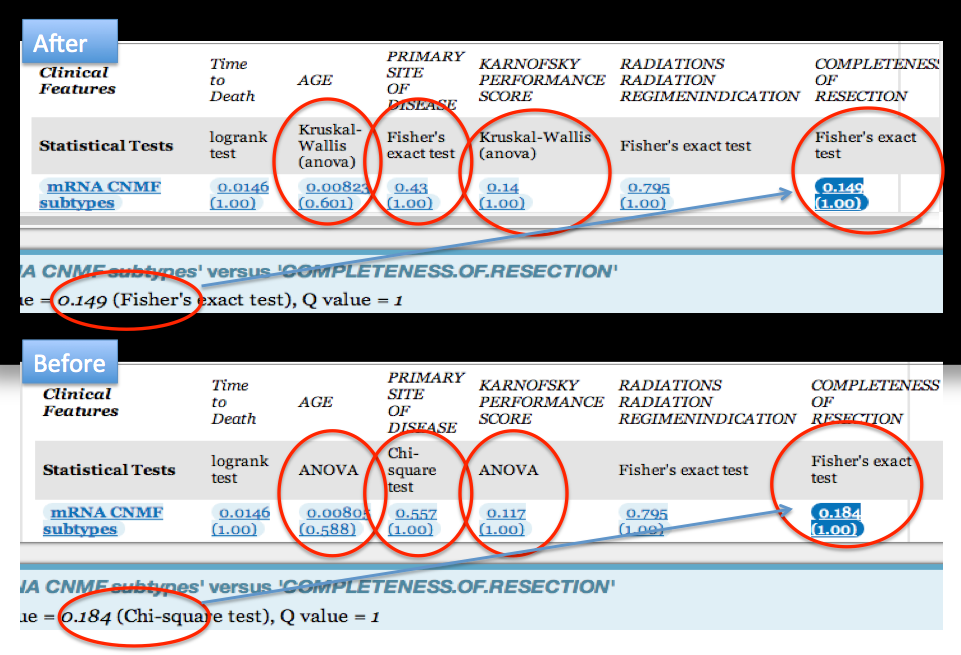
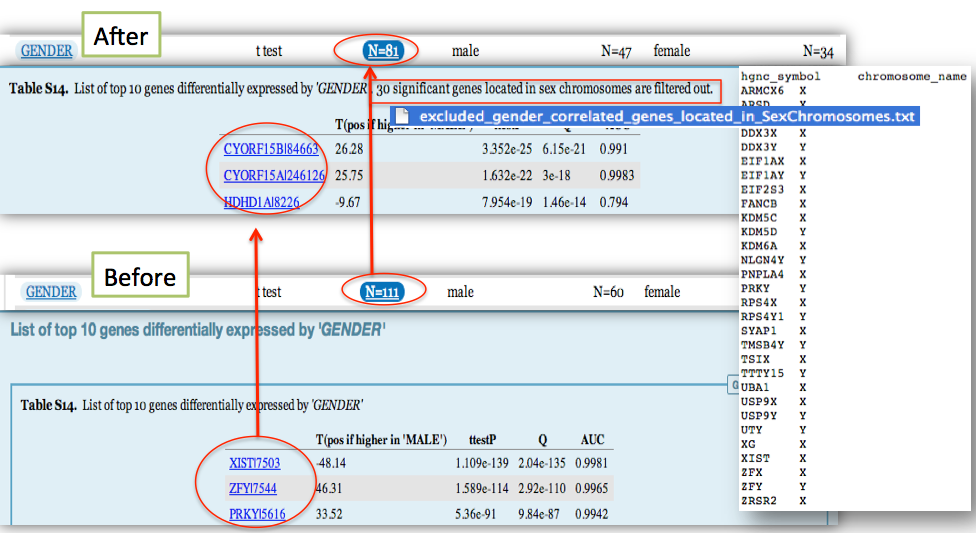
- Significant genes located in sex chromosomes are now filtered, to avoid reporting meaningless correlations
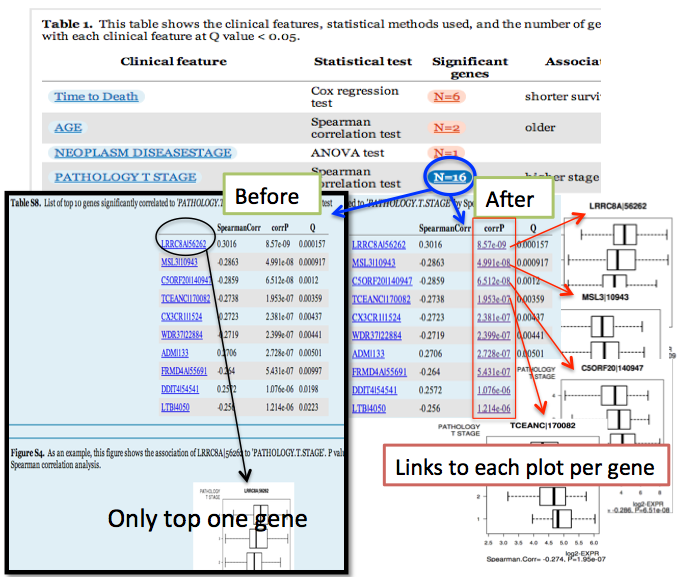
- The box-plot is now given for EVERY one of N top significant genes, instead of only 1 plot for most significant gene
- New Clinical Correlation Analysis: Correlate Clinical vs. Mutation Rate
- Known Issues:
- HotNet was not included, due to a MatLab resource conflict
- CHASM was not included, because it did/would not complete in time
- BRCA: older MutSig versions (1.5 & 2.0) were not run for BRCA cohort: its large size was causing insufficient memory errors
- OV: methylation clustering failed, too few Meth450 samples available but enough that Firehose preferred them over the 592 old Meth27 samples
- Copy Number Analyses:
- GISTIC2 updated to v2.0.21 in our SNP6 and LowPass Copy Number Analyses
Cytoband labels for peaks listed in amp_genes and del_genes outputs use center of maximal segment rather than start of wide peak region (consistent with cytobands in all_lesions output)
D.sdesc can now be a row or a column cell array
- CopyNumber_Gistic2 and CopyNumberLowPass_Gistic2: gene.collapse.method changed from mean to extreme as recommended by Andrew Cherniack
- GISTIC2 updated to v2.0.21 in our SNP6 and LowPass Copy Number Analyses
- Improvements to website:
- Unified stddata and Analyses dashboards into single, cleaner view
- Samples popup for quicklook browsing of sample counts, linked to full samples report and Nozzle analyses reports for those samples
- Clickable Analyses workflow graph: navigate to Nozzle reports for Analyses tasks, directly from the graphical representation of the workflow
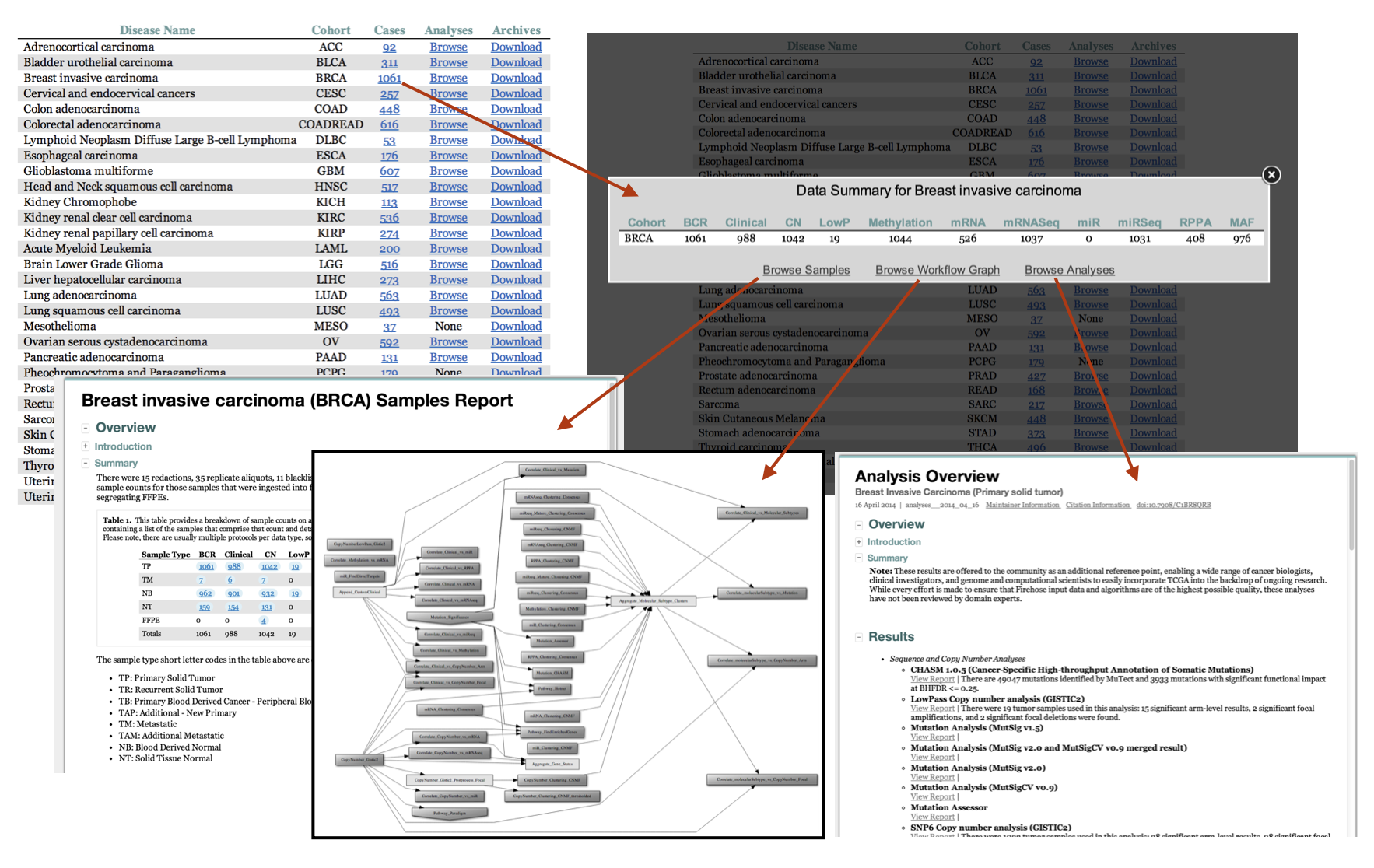
- Number of analyses reports increased to 988
- MutSig nozzle reports updated to include blacklist name
- Preprocessing pipelines for Methylation, miRseq, and mRNA moved from Analysis run to stddata workflow (as noted in April 2014 stddata run)
- CNMF clustering migrated away from GenePattern into Firehose native jobs
- Correlate_Clinical_vs_Molecular_Subtypes results added to all cohorts except PANCAN12
- Mutation_CHASM results added to:
- HNSC
- COAD
- mRNA_Clustering_CNMF result added to PANCAN12
- These additions will be evident in both firehose_get and the online reports
Summary of sample changes since Sept 2013 analysis run (see the comprehensive samples report for more details):
BCR +719 (9267 total) Clinical +245 (7580 total) CN +521 (8487 total) LowP +232 (1091 total) MAF +667 (5335 total) Methylation +489 (8514 total) miR +74 (1135 total) miRSeq +306 (7470 total) mRNA -2 (2217 total) mRNASeq +588 (7478 total) RPPA +1 (4033 total) - Reports:
- Analysis Reports increased in number from 886 to 932
- Samples Report now loads much faster: bulky sections (Filtered Samples, FFPE Cases, and Additional Annotations) converted to links
- Mutation Analyses:
- MutSig reports no longer contain the obsolete clustered mutations section
- MutSig blacklist updated, which helped reduce # of non-signficant genes being mislabeled as significant (see GAF discussion from Sept 2013)
- CHASM analysis report updated to include version number
- CHASM did not complete in time for 9 tumor cohorts
- Copy Number Analyses:
- GISTIC updated to v2.0.20 in our SNP6 and LowPass Copy Number Analyses
- Fixes hang in peak arbitration when all chromosomes (including Y) are present
- Test combinations of res, alpha parameters and maximum copy level that could overflow memory by using too many bins in scoring
- GISTIC output files orig_stats.mat and D.cap1.5.mat now included in results archive, for external use in HotNet and other pathway analyses
- New CopyNumber_Clustering_CNMF_thresholded task added
- Clinical and Correlative Analyses: updated to promote code reuse and unify our AWG and quarterly analyses into a single workflow
- Correlate_Clinical_vs_Molecular_Subtypes
- Correlate_Clinical_vs_miRseq
- Correlate_Clinical_vs_mRNAseq
- Correlate_Clinical_vs_Mutation
- Correlate_Clinical_vs_CopyNumber_Arm
- Correlate_Clinical_vs_CopyNumber_Focal
- Correlate_Clinical_vs_RPPA
- Correlate_Clinical_vs_miR
- Correlate_Clinical_vs_mRNA
- Correlate_Clinical_vs_Methylation
- Correlate_molecularSubtype_vs_CopyNumber_Arm
- Correlate_molecularSubtype_vs_Mutation
- Correlate_molecularSubtype_vs_CopyNumber_Focal
- Aggregate_Molecular_Subtype_Clusters
- Methylation_Preprocess
- mRNASeq Analyses: updated to merge Illumina GA2 and HiSeq data if both are available in similar formats (either RPKM or RSEM)
- Our previous approach (of not merging) was unnecessarily conservative, because samples are typically NOT sequenced on both platforms
- For example: patients A, B sequenced only on Illumina GA2, and patients Y, Z sequenced only on Illumina HiSeq
- Previously, Illumina HiSeq data was used exclusively in downstream analyses when available (RSEM preferred over RPKM).
- This merge should be evident as significantly higher sample counts in downstream mRNASeq analyses (and reports)
- This was previously discussed in the January 2014 Standardized Data Run Release Notes
Summary of sample changes (see the comprehensive samples report for more details):
BCR
+281
(8548 total)
Clinical
+804
(7335 total)
CN
+563
(7966 total)
LowP
-12
(859 total)
MAF
+263
(4668 total)
Methylation
+759
(8025 total)
miRseq
+842
(7164 total)
mRNAseq
+1072
(6890 total)
RPPA
+797
(4032 total)
- Introduced CHASM Analyses for 10 disease cohorts:
- Machine learning method to distinguish between driver and passenger somatic missense mutations
- Driver mutations are curated from the COSMIC database
- Passenger mutations are based on background base substitution frequencies observed for the specific tumor type
- Introduced GISTIC2 analyses for each of the 13 disease studies containing Low-Pass Copy Number data (cna__illuminahiseq_dnaseqc__hms_harvard_edu)
- Mutation Assessor:
- Primary script updated such that runtime is vastly decreased for large MAF files
- New report added summarizing the functional impact of missense mutations at the gene level
- GISTIC2 updated to v2.0.19 to fix minor bugs
- Limit absurdly high CN values to +/- 1e6
- Fix line numbers reported when segment shortened to 0 markers
- Allow single- and zero-marker arms for broad analysis
- Analysis Reports:
- Increased in number to 886
- Enhanced download section of every report to note that firehose_get can be used, or Broad or TCGA sites (including URLs to each)
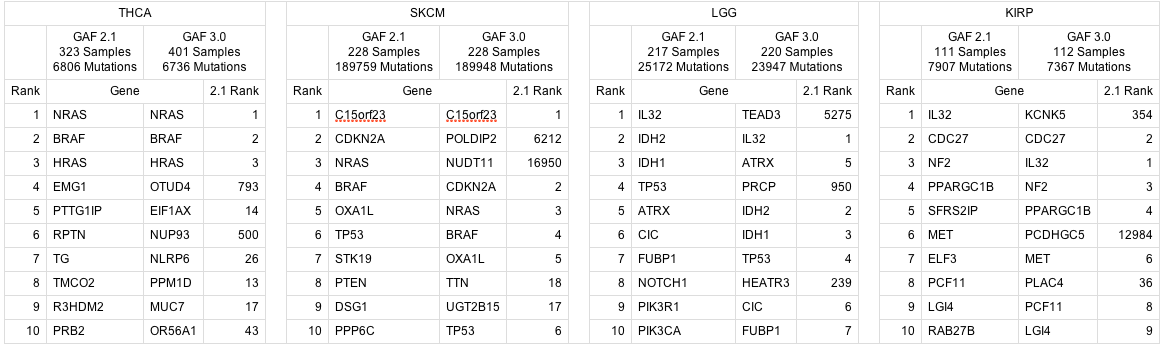
IMPORTANT: issues with gene annotation in GAF 3.0 have impacted mutation analyses in this run. The GAF 3.0 annotation issue has been discussed in the TCGA sequencing working group and corrective action is underway, but until then we strongly advise caution in the interpretation of mutation analyses based upon GAF 3.0. The following mutation analyses are affected in this run
for the following 6 disease studies (which presently have MAFs with GAF 3.0 annotations deposited at the TCGA DCC): KICH, KIRP, LGG, PAAD, SKCM, THCA
Rather than completely remove the affected analyses, we felt it would be valuable diagnostically to have these mutation results available for inspection. Towards that end, here is a comparison of the top 10 significant genes found by MutSig for disease studies that used a GAF 2.1 MAF in the last analyses run (2013_05_23), versus a GAF 3.0 MAF in this run:
- IMPORTANT: this will be the final installment of our monthly cycle of Firehose analyses results. In order to better focus on serving AWG needs, evolving computational methods, and the portals used to disseminate & explore them, we are switching to a quarterly release cycle. Our next Firehose analysis run will therefore be released in September 2013.
Summary of sample changes (see the comprehensive samples report for more details):
BCR
+424
(8267 total)
Clinical
+133
(6531 total)
CN
+208
(7403 total)
LowP
+235
(871 total)
MAF
+116
(4405 total)
Methylation
+120
(7266 total)
miRseq
+102
(6322 total)
mRNAseq
+162
(5818 total)
- Analysis Reports:
- Increased from 690 to 774
- Moved Clinical correlations near top of aggregate reports, to promote greater visibility for potential therapeutic discovery
- Corrected outgoing URLs in clinical reports, to remove
__<number> and |<number>suffixes from gene or miR names; these suffixes are added during analysis to uniquely identify each instance when a given gene/miR appears multiple times (e.g. for more than 1 methylation probe), but having them in URL was breaking lookups in GeneCards or miRbase - Simpler navigation to similar analysis results across disease cohort types, which is especially useful for comparing subtype results in AWG runs:

- New or updated analyses:
- Pathway_Hotnet:
- Parallelized HotNet task, improving performance by factor 3-5X, which should shrink turnaround time of future analysis runs
- We are in ongoing discussions with Raphael Lab at Brown to converge our analyses results with theirs, using THCA as testbed
- miRseq_Mature_Clustering:
- Clustering of miRseq mature expression
- LegoPlotter:
- Generate Lego plots for MutSig generated MAF files
- MergeMutSigRunResults:
- Merge outputs of MutSig 2.0 and MutSigCV for both end-users and downstream analyses
- PARADIGM
- Updated reports to include significance calculation
- Pathway_Hotnet:
- Custom AWG runs since last posted analysis snapshot run:
- Thyroid 2013_06_06 (preliminary to workshop)
- Thyroid 2013_06_20 (for analysis workshop)
- Stomach 2013_06_18
- Melanoma 2013_05_02
- Released firehose_get version 0.3.13: now with 15% kinder messages than before, enhanced the online documentation and examples, and new
-onlyoption
Summary of sample changes: comprehensive samples report is available here (as described previously in our stddata run notes)
BCR
+43
(7843 total)
Clinical
+158
(6398 total)
CN
-30
(7195 total)
MAF
+89
(4289 total)
Methylation
-49
(7146 total)
miRseq
+101
(6220 total)
mRNAseq
+328
(5656 total)
RPPA
+62
(3235 total)
- Search added to GDAC site: explore results more quickly than ever, streamlining the extraction of meaning from TCGA data & Firehose analyses

- Significant enhancements to the structure & layout of our Nozzle reports:
- Introduce a subtitle area, to make the primary title cleaner, while using a smaller font to enable more meta info in report
- Clearly articulate the Firehose run version stamp in subtitle
- Collapse maintainer and citation information within clickable dropdown boxes of subtitle
- Added Digital object identifiers (DOIs), thus enabling over 700 reports we generate per month to be citable directly in the literature

- Updated tasks, with a collection of small improvements to analyses, figures and report narratives:
- Correlate_Clinical_vs_Molecular_Subtypes (renamed Signatures -> Subtypes)
- Correlate_Clinical_vs_CopyNumber_Arm
- Correlate_Clinical_vs_CopyNumber_Focal
- Correlate_CopyNumber_vs_miR
- Correlate_Methylation_vs_mRNA
- Correlate_molecularSubtype_vs_CopyNumber_Ar
- Correlate_molecularSubtype_vs_CopyNumber_Focal
- RPPA_Clustering_CNMF
- RPPA_Clustering_Consensus
- Improved aesthetics and layout of home page
Sample Changes: more than 1600 new aliquots ingested
BCR
+340
(7800 total)
Clinical
+133
(6240 total)
CN
+369
(7225 total)
Methylation
+436
(7195 total)
miRseq
+59
(6119 total)
mRNA
-3
(2219 total)
mRNAseq
+310
(5328 total)
- 4 custom runs for AWGs, including several with multiple subtypes: those with active links are available via firehose_get and the respective dashboards:
- awg_hnsc__2013_03_30
- AWG_LGG_2013_04_06
- AWG_THCA__2013_03_18
- awg_stad__2013_04_17
- Minor organizational & documentation enhancements to GDAC site and dashboards
- MutSig S2N is deprecated and has been removed from pipeline. It has been replaced with MutSig CV. MutSig 1.5 has also been added.
- CoMut for MutSig2.0 has been corrected to use a Q threshold of 0.1
- The 2013_02_22 analyses run for CoMut mistakenly used a threshold of 0.5
- Added directed graph of tasks in each analysis run, to both our FAQ and documentation pages. This will be enhanced in the near future, with a version that allows one to click on a node in the graph to determine inputs for that pipeline task, link to its Nozzle report, etc.
- Incomplete Pipelines:
- CoMutCV failed for LAML
- HotNet did not complete before release for:
- COADREAD
- BRCA
- SKCM
- A parallelized version of the HotNet task is under development, and expected to run 3-5 times faster. This will shrink our monthly turnaround times.
- Updated Pipelines:
- miRseq_Clustering_Consensus
- miRseq_Clustering_CNMF
- CopyNumber_Gistic2_Postprocess_Focal
- Correlate_Clinical_vs_CopyNumber_Focal
- Correlate_Clinical_vs_CopyNumber_Arm
- CopyNumber_Clustering_CNMF
{kind=link}
Sample changes since last analyses run
BCR
-8
(7460 total)
Clinical
+158
(6107 total)
CN
+304
(6856 total)
LowP
+35
(636 total)
MAF
+266
(4200 total)
Methylation
+141
(6759 total)
miRseq
+433
(6060 total)
mRNAseq
+228
(5018 total)
- Increased number of output reports from 614 to 703
- New AWG Analyses:
- New pages added on NCI wiki and Broad GDAC site to describe and reflect custom Firehose runs for TCGA analysis working groups
- Performed and/or released custom AWG runs for LGG, LUAD, GBM, PANCAN12, and THCA
- New Correlation Analyses (see Nozzle reports in dashboard for more info):
- Correlate_molecularSubtype_vs_CopyNumber_Arm
- Correlate_molecularSubtype_vs_CopyNumber_Focal
- Correlate_molecularSubtype_vs_Mutation
- Mutation Analyses:
- CoMut plot inconsistency for MutSig 2.0 results
- The CoMut plot was generated using a Q threshold of 0.5 (0.1 was used in previous runs)
- Many more genes are displayed in the plot due to this change
- This change was inadvertent and the Q threshold will be reverted to 0.1 in the next analyses run
- MutSigCV introduced (MutSig2N is deprecated, and will be removed next month)
- MutSig is now compatible with WashU WIG files
- MutSigPreprocess now more configurable has Firehose native task (no longer GenePattern)
- Bug fixed that only manifests in certain cases, and only when there are WGS+capture MAFs for some of the same patients
- GenerateStickFigures* DCC submission archive removed: its content rolled into general Mutsig DCC archive
- CoMut plot inconsistency for MutSig 2.0 results
- Copy Number Analyses
- GISTIC now more configurable as Firehose native task (no longer GenePattern)
- Updated to version 2.0.17a:
- memory and performance optimization of peak identification code.
- SegArray version 1.06 with Mex files added to improve performance.
- Fix gistic_plots chromosome shading for q-value 0.
- fix bug in 2.0.17 where output file "raw_copy_number.pdf" was being named "[pathname '.pdf']"
- CopyNumber_GeneBySample removed (was only for hg18; its output has been superseded by GISTIC)
- Pathway Analyses:
- Now in their own "Pathway Analyses" section of top-level aggregate reports, instead of catch-all "Other" section
- Restored clarity of pathway analysis task names & output archives:
- Pathway_Paradigm_mRNA
- Pathway_Paradigm_mRNA_And_Copy_Number
- Pathway_Paradigm_RNASeq
- Pathway_Paradigm_RNASeq_And_Copy_Number
- Pathway_Hotnet
- This changes the name of their respective DCC submission archives
- Output of cluster aggregator Aggregate_Molecular_Subtype_Clusters now packaged for DCC submission:
- Provides a table of patient vs. clusterings (per datatype, per clustering method)
- Where each cell indicates the respective patient membership in that given cluster group
- General Software Tools:
- firehose_get v0.3.11 released
- The internal /wiki/spaces/GDAC/pages/844333857 tool can now input a custom sample set file, to facilitates efficiently analyzing cancer subtypes
Sample changes since last analyses run:
BCR
+161
(7468 total)
Clinical
+40
(5949 total)
CN
+122
(6552 total)
MAF
+136
(3934 total)
Methylation
+96
(6618 total)
mRNA
-2
(2222 total)
mRNAseq
+110
(4790 total)
- PARADIGM mRNASeq input now being median centered using normals if they exist
- New pipeline added: HotNet analyses on 18 disease cohorts
- Analysis Reports:
Increased in number from 551 to 614
Now reflect the diease cohort over which the analysis was performed: e.g. primary solid tumor, metastatic, subtype designations (like astrocytoma), etc
- LGG MAF-swap issue corrected (see Standardized Data Run Release Notes for 21 Dec. 2012)
- Sample changes since last analysis run:

- New mutation analyses (and MAFs) for: HNSC, KIRP, LGG, PAAD, SKCM
- Updated mutation analyses (and MAFs) for: GBM and KIRC
- New PANCAN12 and PANCAN18 cohorts, with a small handful of analyses
- 2 new kinds of Paradigm runs, against RNASeq and RNASeqWithCopyNumber
- Correlations against several new clinical CDEs, including smoking-related (where applicable). For more details see the stddata notes at
Standardized Data Run Release Notes
- Avoid over-interpretation of survival events in the presence of low-count statistics, by assigning P value of 100; this prevents clinical correlations from interpreting such events as significant when generating survival curves.
- Due to an internal configuration error, LAML analyses were reduced from 26 to 12. This will be corrected in the next run.
- More than 3000 new samples ingested, reflected in a total of 475 analysis reports generated.
Sample differences:
BCR +171 (7123 total) Clinical +119 (5798 total) CN +398 (6212 total) LowP +23 (501 total) Methylation +882 (6471 total) mRNA +1 (2225 total) mRNAseq +808 (4371 total) miRseq +851 (5644 total) MAF +323 (3183 total)
- Internally this was a big release, because every pipeline was rewired to accomodate using sample sets of tumors, or normals, or type-specific subsets of each
- But that should in principle be transparent to external users: i.e. normals added as of stddata__2012_10_24 are NOT YET REFLECTED in Firehose analyses (because very few analyses employ them at present)
- MutSig (v2.0) has been updated to restore the clustered mutations result
- Correlate_Methylation_vs_mRNA task was fixed to restore probe information removed in previous run
- Two enhancements to samples summary report available on our dashboard
- Now lists every sample that is filtered from the datastream, with an explanation of why (see stddata__2012_10_24 release notes for more details)
- Heatmaps are now included that display available samples per data type vs. participants
- Now lists every sample that is filtered from the datastream, with an explanation of why (see stddata__2012_10_24 release notes for more details)
- firehose_get v0.3.8: support additional awg runs, such as awg_thca__2012_10_24 and awg_luad__2012_11_15
Sample Changes
BCR
+71
(6952 total)
Clinical
+8
(5679 total)
CN
+4
(5814 total)
Methylation
+118
(5589 total)
miRseq
+684
(4793 total)
mRNAseq
+36
(3563 total)
RPPA
+442
(3173 total)
- Updated Pipelines:
- Aggregate_Clusters & Correlate_Clinical_vs_Molecular_Signatures
- Now handle copy number cNMF and RPPA clustering results.
- Now handle copy number cNMF and RPPA clustering results.
- Mutation_Assessor
- Now handles headers as per the MAF Spec
- Methylation
- Major overhaul and optimizations added to improve clustering and expression correlation results.
- Aggregate_Clusters & Correlate_Clinical_vs_Molecular_Signatures
- MutSig
- Changed from GenePattern pipeline to Firehose workflow:
- yielding more transparency & job-avoidance on intermediate processing steps
- and changed output archive names from gdac*Mutation_Significance*
- to MutSigPreprocess2.0, ProcessCoverageForMutSig2.0, MutSigRun2.0, MutSigNozzleReport2.0, MutSigNozzleReportS2N
- Current version of MutSig (v2.0) is not computing a clustered mutations result. This will be fixed in the next analysis run.
- New version of MutSig (S2N) added to our analysis run
- Please note that this version is still in active development and as a result, the significant genes list contains false positives. The algorithm will continue to be refined to eliminate these false positives. In the meantime, an intersection of significant genes found by this version and MutSig v2.0 is suggested to eliminate false positives.
- Please note that this version is still in active development and as a result, the significant genes list contains false positives. The algorithm will continue to be refined to eliminate these false positives. In the meantime, an intersection of significant genes found by this version and MutSig v2.0 is suggested to eliminate false positives.
- Changed from GenePattern pipeline to Firehose workflow:
- firehose_get v0.3.7: reflect addition of PANCAN8 disease cohort, and resumption of COAD and READ cohorts (on top of existing COADREAD aggregate)
Correlate_Clinical_vs_Methylation currently runs on one methylation probe per gene chosen by negative correlation with corresponding expression data (mRNA/mRNAseq). When insufficient expression data exists, methylation correlation pipelines do not run. In future iterations, when there is insufficient expression data, Correlate_Clinical_vs_Methylation will run on mean probe values per gene.
- The past 5 months of Standardized Data have been loaded into IGV:
- Partitioned by reference genome - When choosing "Load from Server...", only the data for the currently selected reference (Human hg18/Human hg19) will be available via the menu.
- Copy Number data available for both hg18 and hg19, both with and without germline samples
- Meth450 data now available
To acces, open IGV, and with Human hg18/hg19 selected as the reference, navigate:
File -> Load from Server... -> The Cancer Genome Atlas -> TCGA Broad GDAC -> Firehose Standard Data
- New Pipelines:
- Aggregate_Gene_Status
- CopyNumber_Clustering_CNMF
- CopyNumber_Gistic2_Postprocess_Focal
- Correlate_Clinical_vs_Methylation
- Correlate_Clinical_vs_RPPA
- Correlate_Clinical_vs_mRNAseq
- Correlate_Clinical_vs_miRseq
- Updated Pipelines:
- Methylation
- Changed how gene-level data is selected from probe-level data. We group probes by gene, and then:
- For clustering: select the probe with the greatest stddev across all individuals.
- For correlation: for each individual, select the probe with the median signal.
- Changed which mRNA expression data is used for correlation:
- meth27 is still correlated against mRNA.
- meth450 is now correlated against mRNAseq.
- Changed how gene-level data is selected from probe-level data. We group probes by gene, and then:
- Mutation Assessor
- No longer using web service, now uses correct reference for each MAF rather than hg18 for all of them
- *_Clustering_CNMF
- Corrected issue that caused Nozzle reports to randomly have tables with the first data row as a header
- Methylation
- 111 new reports, for a total of 395 in this run.
- Updated default GISTIC2 parameters - for a description of these parameters, please view the Methods & Data Input Description section of any GISTIC nozzle report created in our 06/23/2012 analyses run or later.
- Old parameters
- Amplification Threshold: 0.3
- Deletion Threshold: 0.3
- Cap Values: 2.0
- Broad Length Cutoff: 0.5
- Remove X-Chromosome: True
- Join Segment Size: 10
- Maximum Sample Segments: 10000
- Updated parameters
- Amplification Threshold: 0.1
- Deletion Threshold: 0.1
- Cap Values: 1.5
- Broad Length Cutoff: 0.7
- Remove X-Chromosome: False
- Join Segment Size: 4
- Maximum Sample Segments: 2000
- Old parameters
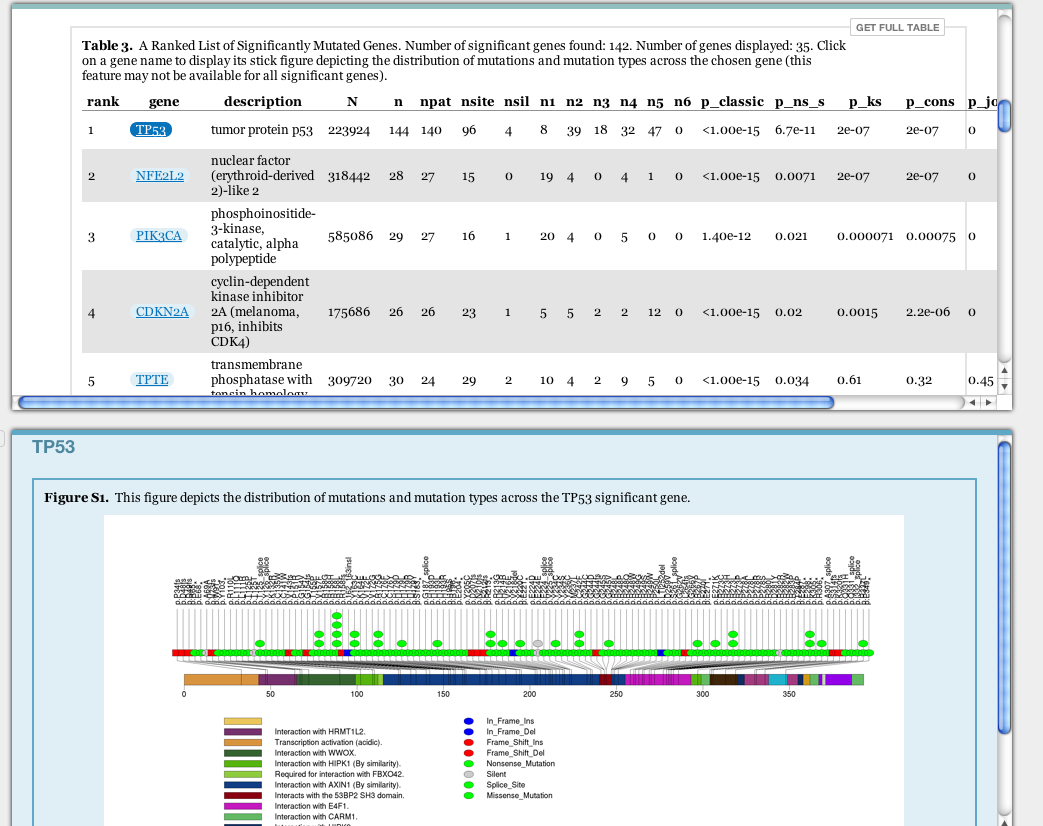
- MutSig nozzle report updated to include stick figures depicting the distribution of mutations and mutation types across each significant gene. Click on any significant gene that appears as a link to display the figure. This feature may not be available for every gene for 2 reasons: (1) a lack of required information for the particular significant gene or (2) lack of required columns in the input MAF.
- 102 COADREAD samples missing from prior Mutation Significance run have been re-included. See our email archive for an explanation of this issue.
- firehose_get v0.3.5:
- tweak date regex to correctly detect October months
- support downloading arbitrary run types, not just stddata or analyses (e.g. awg_pancan8)
- allow ~ in -task option to connote that given task(s) should be EXCLUDEd
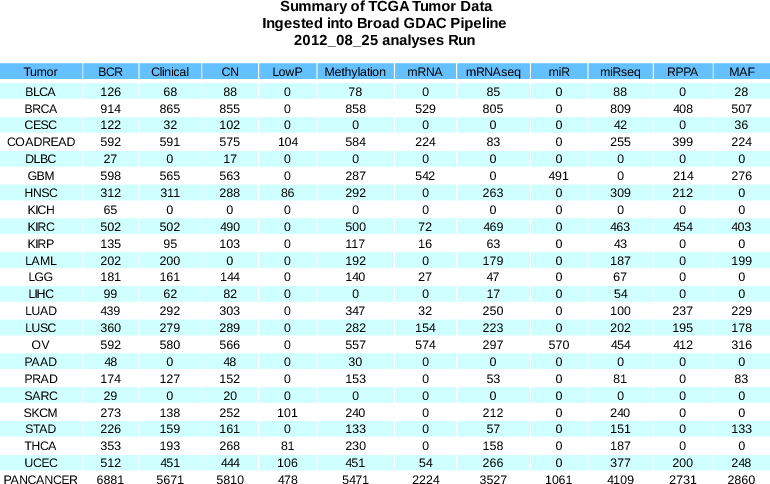
Sample Changes:
BCR
+35
(6881 total)
Clinical
+38
(5671 total)
CN
+424
(5810 total)
LowP
+402
(478 total)
Methylation
+6
(5471 total)
miR
+6
(1061 total)
miRseq
+133
(4109 total)
mRNA
+6
(2224 total)
mRNAseq
+67
(3527 total)
RPPA
+644
(2731 total)
New Samples:
BCR +160 (6846 total) Clinical +55 (5633 total) Methylation +140 (5465 total) mRNAseq +306 (3460 total) miRseq +195 (3976 total) - 32 new reports, for a total of 284 in this run
- New Pipelines:
- Correlate_Clinical_vs_CopyNumber_Focal
- Correlate_Clinical_vs_CopyNumber_Arm
- MutSig:
- To address item (9) from 2012_06_23 Analysis Run Release Notes, input WIGs (from stddata__2012_07_25) updated to hg19 for:
- BLCA
- BRCA
- CESC
- KIRC
- LUSC
- LUAD
- PRAD
- STAD
- UCEC
- STAD failing due to memory exhaustion - ignoring for this release as there is no currently active AWG
- COADREAD analysis is missing 102 samples. Please see explanation in our email archive.
- To address item (9) from 2012_06_23 Analysis Run Release Notes, input WIGs (from stddata__2012_07_25) updated to hg19 for:
- Gistic2
- Failed for PANCANCER due to memory exhaustion; ignored because it is the large 23-tumor cohort, not yet the 8-tumor cohort defined by AWG.
- Failed for PANCANCER due to memory exhaustion; ignored because it is the large 23-tumor cohort, not yet the 8-tumor cohort defined by AWG.
Increased number of archives generated from 777 to 993
- Increased number of reports from 227 to 252
- 2,244 new samples reflected since May analyses run, due to more data and better counting:
- 76 LowP (new sample type - Low Pass DNAseq)
- 230 BCR
- 307 Clinical
- 618 mRNAseq
- 937 miRseq
- 76 MAF
- GISTIC2 report now includes a description of both the input and output files in the Methods & Data section
Methylation data:
Rewired pipelines to include meth450 platform, and also give it preference over meth27 when both are present.
(Methods to combine 450 & 27 analytically are not in Firehose: would be nice for AWGs to provide if possible)This greatly increases count of methylation samples flowing through analyses (e.g. UCEC 117–>363)
- Most clusterings show similar results, but some are discordant with previous runs: we could use AWG help to evaluate, and will post comparative analysis online towards that end
- New clustering pipelines heuristic: a sample will be dropped from analyses when 80% or more genes are absent.
- mRNAseq: we now utilize rnaseqv2 archives, but fall back to v1 rnaseq when v2 is not available for a given tumor type
- RSEM estimation used for downstream clustering & correlation analysis, when available, otherwise RPKM estimation will used
- RSEM is used to estimate gene and transcript abundances (http://deweylab.biostat.wisc.edu/rsem/rsem-calculate-expression.html); values are normalized to a fixed upper quartile value of 1000 for gene and 300 for transcript level estimates, and the normalized values are placed in a separate file (From the DCC document).
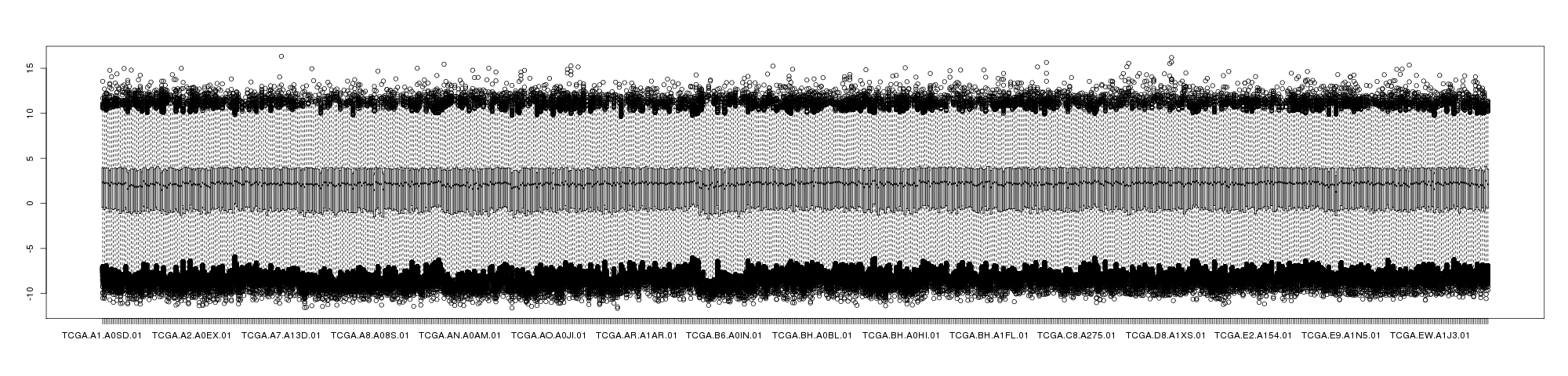
The following showed the boxplot of BRCA mRNAseq samples with log2 transformed RESM (left) and RPKM (right).

- Improvements to clinical correlations:
- Use try/catch to avoid needless failures when parameters are moved in XML scheme
- Towards aim of having survival curves ALWAYS generated for ALL disease types
- Archives now generated for clinical correlations: 33 in this run alone (versus zero in previous runs)
- MutSig:
- Updated to v2.0, which among other enhancements now distinguishes between hg18 and hg19 builds. Alas, we did not correct for hg19 in time for the run, so results for all tumor data based upon hg19 (BLCA, BRCA, CESC, KIRC, LUSC, LUAD, PRAD, STAD, and UCEC) SHOULD NOT BE USED; archives were not posted to DCC, but the reports will remain online for inspection.
- Nozzle report now includes visually compelling, integrative"CoMut" plot.
- Increased number of pipelines in workflow from 27 to 33, adding:
- Correlate_Clinical_vs_Molecular_Signatures
- Correlate_Clinical_vs_mRNA
- Correlate_Clinical_vs_Mutation
- Correlate_Clinical_vs_miR
- Correlate_CopyNumber_vs_mRNAseq
RPPA_Clustering_CNMF
RPPA_Clustering_Consensus
- Paradigm_Lite
- Increased number of analysis output reports from 167 to 227
- Increased number of tumorsets with 100% success from 12 to 17
As noted in the 2012_05_15 data run, Broad GCC SNP6 data has been reprocessed from the single platform
into 4 distinct platforms (each containing the full complement of samples):
Merge_snp__genome_wide_snp_6__broad_mit_edu__Level_3__segmented_scna_hg18__seg
Merge_snp__genome_wide_snp_6__broad_mit_edu__Level_3__segmented_scna_hg19__seg
Merge_snp__genome_wide_snp_6__broad_mit_edu__Level_3__segmented_scna_minus_germline_cnv_hg18__seg
Merge_snp__genome_wide_snp_6__broad_mit_edu__Level_3__segmented_scna_minus_germline_cnv_hg19__seg
Gistic in our GDAC pipeline now operates upon the last of these: scna_minus_germline_cnv_hg19__seg- Internal support for analyses of meth450 platform is underway but incomplete: the data have increased more than an order of magnitude in size, which necessitated algorithmic refactoring and additional testing. Look for meth450-based analyses in June.
- Mutation significance & assessor results for lung cancer (LUSC and LUAD tumor types) continue to be removed, as described below.
- Doubled our 100% success rate by eliminating vacuous failures due to having zero data of given type available.
- 3 new analyses performed (with output reports) for BLCA: MutSig, and CNMF and consensus clustering for mRNAseq
- Paradigm output file merge_merged.tab renamed to InferredPathwayLevels.tab to more accurately identify its contents.
- RNA-Seq preprocessing pipeline results now packaged for DCC upload
- Intermediate results of expression clustering pipelines now packaged for DCC upload
- GISTIC inputs listed in report and reference files included in results archive.
- Mutation significance results for lung cancer (LUSC and LUAD tumor types) continue to be removed, as described below.
- Over 2000 new Methylation samples ingested: with caveat that we do not yet merge multiple platforms (e.g. humanMethylation27 with 450), so methylation analyses will still refect only 1 platform and thus lower counts
- New MAFs: BLCA (28 samples), CESC (36), and STAD (133)
- Initial Mutation_Signficance analyses and Mutation_Assessor annotations for CESC and STAD
- Mutation assessor module updated to reflect newer web service version and correct Mutation_Status column etc in outputs
- Reflect individual RNA-Seq expression clustering reports in top-level aggregate report
- New firehose_get utility, for simpler downloads
- Mutation significance results for lung cancer (LUSC and LUAD tumor types) continue to be removed, as described below
- As noted in the stddata run release notes, nearly 2000 new samples over multiple data types.
- Revamped dashboard table layout, to more clearly indicate which links are to public analysis reports and which lead to credentialed DCC downloads.
- Mutation significance results for lung cancer (LUSC and LUAD tumor types) continue to be removed, as described below.
Major step forward in accessibility, by linking our analysis reports for each tumor type run directly to our WWW dashboard, sans login credentials; alone, this run contains 138 reports, each accessible by a single web-click. We hope this furthers the TCGA mission by lowering the entry barrier to use, thereby helping to broaden the impact of TCGA data and analyses.
Six new analysis pipelines:
mRNAseq_Clustering_CNMF
mRNAseq_Clustering_Consensus
miRseq_Clustering_CNMF
miRseq_Clustering_Consensus
Pathway_Paradigm_Expression
Pathway_Paradigm_Expression_CopyNumber
The latter two employ the full/scatter-gather version of Paradigm; the previous Pathway_Paradigm version has been renamed to Pathway_Paradigm_Lite, and will continue to be run in our Analyses workflow until all are satisfied it can be deprecated.
To simplify data retrieval, the Tumor Type cells in our dashboard now link directly to the DCC locations from which the corresponding archives may be downloaded.
- MutSig results for LUSC and LUAD are removed from this run, due to a discrepancy in the set of genes identified as most significant by Mutsig v1.5 versus the set manually curated by the TCGA lung AWG. New LUSC and LUAC MutSig results will be available once MutSig is upgraded to v2.0 in the Broad GDAC pipeline.