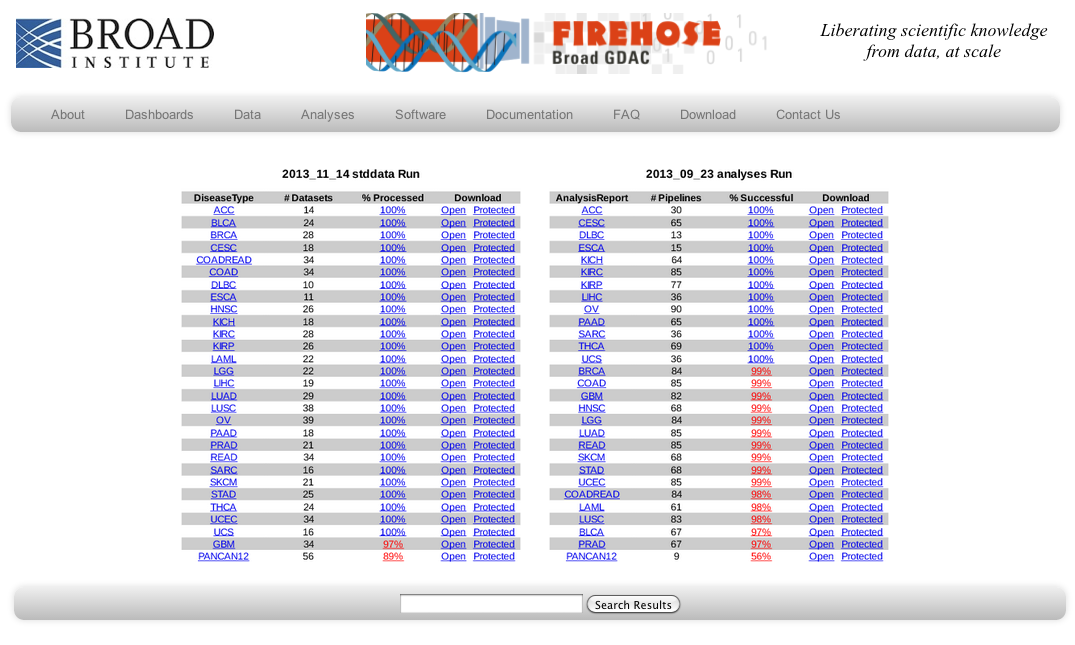
Standardized Data Run Release Notes
- Former user (Deleted)
- Former user (Deleted)
- Former user (Deleted)
The LIHC RPPA data submitted by MDACC were discovered to be mislabelled MESO samples. The analyses and standard data pipelines for LIHC have been re-run using the corrected samples submitted in March, and the nozzle reports now contain notices of the discrepancies.
The following table shows the changes in sample counts for RPPA data as a result of this patch:
HNSC | +145 | (357 total) |
|---|---|---|
LIHC | +121 | (184 total) |
THCA | +146 | (368 total) |
Summary of sample changes (see the comprehensive samples report for more details):
Clinical
+8
(11196 total)
MAF
+122
(7099 total)
mRNASeq
+214
(10267 total)
rawMAF
+1576
(6322 total)
RPPA
+626
(7429 total)
- Significant changes to clinical data processing, to accommodate new XSD 2.7 format adopted by TCGA:
- Per TCGA this new format is not backwards compatible with the previous XML, so if you use a parser customized to the previous XSD, it may not work correctly. This change is confined to the XML, no other TCGA data is affected by this change.
- These changes are detailed at https://wiki.nci.nih.gov/display/TCGA/Release+1.39.1#Release1.39.1-XSD2.7Implementation
- In the new format, 'CQCF' parameters are removed. Instead, they are provided in new XML format, SSF.
- According to this change, 'cqcf' parameters are removed and "patient.tumor_samples" and "patient.normal_controls" parameters are added in the clinical merged data.
- Added parameters of two new XML formats, OMF and SSF in the clinical merged data
- Fix a bug in Methylation_Preprocess that caused duplicate entries in methylation data
- Added outlinks to TCGA Encyclopedia to the About and Documentation menus of the Broad GDAC Firehose website
- FireBrowse v1.1.24:
- Loaded this data run
- Numerous internal updates to further simplify deployment, which e.g. will help provision AWG-specific databases
- The HeartBeat API function now includes: the directory from and time at which the API server was launched
- Dropped beta clause from version
All mRNASeqV2 packages and derived packages have been updated for MESO and SKCM. Please ensure you are using the following 1.0 archives rather than the 0.0 ones for these cohorts:
- Merge_rnaseqv2__illuminahiseq_rnaseqv2__unc_edu__Level_3__RSEM_genes__data
- Merge_rnaseqv2__illuminahiseq_rnaseqv2__unc_edu__Level_3__RSEM_genes_normalized__data
- Merge_rnaseqv2__illuminahiseq_rnaseqv2__unc_edu__Level_3__RSEM_isoforms__data
- Merge_rnaseqv2__illuminahiseq_rnaseqv2__unc_edu__Level_3__RSEM_isoforms_normalized__data
- Merge_rnaseqv2__illuminahiseq_rnaseqv2__unc_edu__Level_3__exon_quantification__data
- Merge_rnaseqv2__illuminahiseq_rnaseqv2__unc_edu__Level_3__junction_quantification__data
- Methylation_Preprocess
- mRNAseq_Preprocess
This is due to multiple versions of the same protocol in the SDRFs for these cohorts causing our data ingestor to drop samples. After conferring with the submitters, we've updated our tools to make an exception for this with RNASeqV2 data, and added the dropped samples back to the run.
Summary of sample changes (see the comprehensive samples report for more details):
BCR
+1
(11368 total)
Clinical
+24
(11188 total)
CN
+2
(10987 total)
MAF
+191
(6977 total)
Methylation
+1
(10972 total)
miRSeq
+2
(10156 total)
mRNASeq
-50
(10053 total)
rawMAF
+496
(4746 total)
RPPA
+1
(6803 total)
FireBrowse 1.1.21 beta: updated to reflect this data run
Summary of sample changes (see the comprehensive samples report for more details):
BCR
+15
(11367 total)
Clinical
+106
(11164 total)
CN
-2
(10985 total)
LowP
+120
(1211 total)
MAF
+48
(6786 total)
Methylation
+16
(10971 total)
miRSeq
-4
(10154 total)
mRNASeq
+8
(10103 total)
RPPA
+650
(6802 total)
Extensive improvements to clinical data, in that
Clinical_Pick_Tier1archives now bundle 2 forms of values:Entire set of TCGA CDEs, verbatim (in new
All_CDES.txtfile): adding over 700 additional clinical parametersIn addition to the CDE subset normalized by Firehose for downstream analyses (in
<cohort>.clin.merged.picked.txtfile)- For example, to date the ACC picked file has contained less than 20 CDEs while
All_CDEs.txtnow contains more than 100. - Followup values are merged, when available, to yield the most up-to-date values per CDE
- Corrected problem wherein some True/False values for
regimen_indication CDEwere erroneously swapped - Created an interactive table CDEs, which on a single page shows exactly what CDEs are selected for analyses in Firehose for all disease cohorts. Updated the FireBrowse clinical samples API to reference this new CDE table
- Enhanced
Merge_Clinicalpipeline to leverage auxiliary CDEs when available (COAD, READ, ESCA): for all primary CDEs that also have a value in the aux CDE file (e.g. MSI), we now replace the primary value if it is NA and the aux value is not NA
- Extensive improvements to TCGA mutation data:
- New MAF for Diffuse Large B-cell Lymphoma (DLBC, 48 mutation samples)
- Oncotator now included in Firehose mutation pipelines, to standardize TCGA MAFs to a common format:
- This substantially improves the consistency and utility of TCGA MAFs
- hg18 MAFs lifted over to hg19
- All MAFs re-annotated against Gencode v19
- Oft-requested custom columns, such as amino acid change, now present in all MAFs
- Oncotated MAFs are available in 2 pipeline output archives
- Mutation_Packager_Oncotated_Calls
- Mutation_Packager_Oncotated_Raw_Calls
- Extensive improvements to RPPA data, fostering more robust automated processing and downstream analysis:
- Merge_protein_exp__mda_rppa_core__mdanderson_org__Level_3__protein_normalization__data
- Validation and animal source suffixes now stripped off of antibody name to account for several new batches that no longer include them
- Now returns a union of antibody names from all samples, rather than failing when all samples don't have the exact same antibodies
- This does not fix the below RPPA issues when different names for the same antibodies are used (e.g. Acetyl-a-Tubulin-Lys40/Acetyl-a-Tubulin(Lys40), ARHI/DIRAS3) - in these cases each antibody name will appear on separate lines of the merged files until a fix is made at MDACC.
- This enables RPPA analysis for aggregate cohorts such as STES, KIPAN, and GBMLGG
- RPPA_AnnotateWithGene: normalizes the antibody reference files provided by MDACC into a two-column tsv with standardized header, gene name, and antibody name (stripped of suffixes). This file is now provided in the archive.
- Merge_protein_exp__mda_rppa_core__mdanderson_org__Level_3__protein_normalization__data
- FireBrowse v1.1.17 beta:
- Ingest this August 2015 data run:
API through which Firehose-picked and normalized clinical data are accessible has been renamed to Samples/Clinical_FH
Verbatim TCGA clinical data may be accessed through the new Samples/Clincal API
A CDE will be reflected in either API only when it has a value other than NA for at least 1 patient case in any disease cohort.
- For backward compatibility, the Samples/ClinicalTier1 remains available (as a synonym for Samples/Clinical_FH)
- fbget Python and UNIX CLI bindings have been suitably updated
- Which makes it extremely easy to determine for what patients and cohorts any given CDE is defined: e.g.
fbget clinical cde=gleason_score
will show each patient case with a validgleason_score, across all cohorts
- viewGene: now enforces rendering of at most 1 gene per Submit; if multiple genes are given the first is selected
- iCoMut:
- Popup tooltips for mutation panels now show total # of mutations AND fractional % of a given type (e.g. missense)
- New search feature, enabling one to see into which clusters/panels/etc a given patient (or set of patients) falls
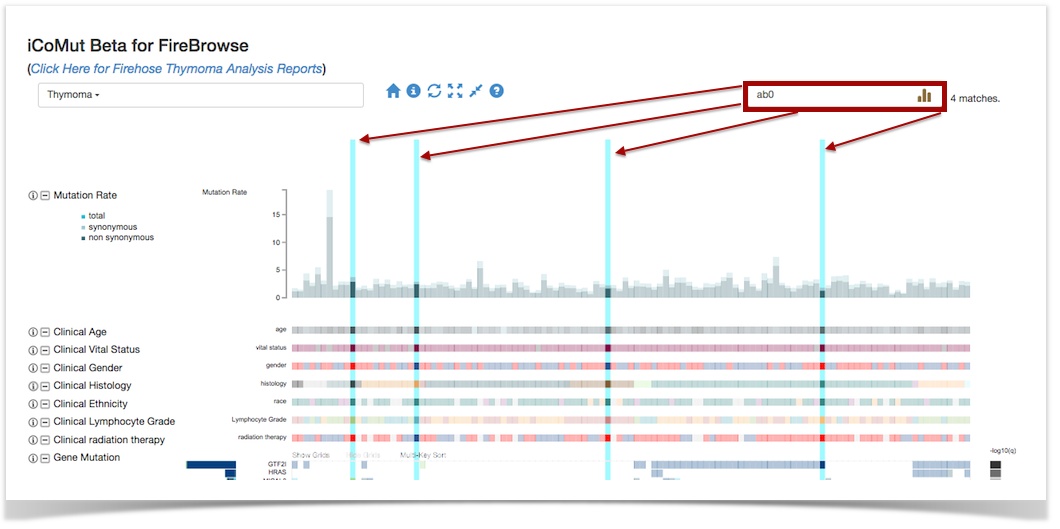
- After extensive testing, upgraded backend database from Mongo2.x to Mongo3.x: v3 dramatically reduces the memory footprint and data storage sizes, which yields greater performance and also clears considerable breathing room to add more data APIs (e.g. for methylation, RPPA, etc) as well as AWG-specific databases
- Ingest this August 2015 data run:
- Corrected item # 9 from Spring 2015 data run: missing RPKM aliqouts in COAD
- Issues:
- RPPA issues due to changes in new data file row names: these have been previously reported to MDACC.
- KIRP: ARHI-M-E -> DIRAS3-M-E (batch 2.0.0)
- LGG: Acetyl-a-Tubulin-Lys40-R-C -> Acetyl-a-Tubulin(Lys40)-R-C (batch 2.0.0)
- PAAD: DIRAS3-M-E -> ARHI-M-E (batch 1.2.0)
- STES: ARHI-M-E -> DIRAS3-M-E (batch 2.0.0)
- RPPA issue with KIRC antibody file:
Gene names were missing for 15 antibodies. The gene names were located in an online supplement file, and manually added:
CA9
CA9
SDHB
Complex-II_subunit30
GYG1
GYG-Glycogenin1
GYS1
GYS
GYS1
GYS_pS641
HIF1A
HIF-1_alpha
LDHA
LDHA
LDHB
LDHB
MTCO2
Mitochondria
ATP5A1
Oxphos-complex-V_subunitb
PKM2
PKM2
PYGB
PYGB
PYGB
PYGB-AB2
PYGL
PYGL
PYGM
PYGM
- RPPA issues due to changes in new data file row names: these have been previously reported to MDACC.
Summary of sample changes (see the comprehensive samples report for more details):
Clinical +113 (11058 total) MAF +154 (6738 total) miRSeq -2 (10158 total) rawMAF +4250 (4250 total) RPPA +696 (6152 total) - FireBrowseupdated to v1.1.11 beta to reflect ingestion of these data
- Python and UNIX fbget wrappers likewise updated to 0.1.4
- New / Updated MAFs:
- SARC (247 samples)
- Curated PRAD MAF: this MAF contains 332 samples, or 93 less than the rawMAF it replaced (and is why the total MAF count has not gone up by 247, but rather 247-93=154).
- rawMAFs: note that these were analyzed in 2015_04_02 analysis run, but heretofore not packaged within a stddata run
- BLCA (395 samples)
- COAD (367 samples)
- COADREAD (489 sample)
- GBMLGG (803 samples)
- HNSC (508 samples)
- KIPAN (678 samples)
- KIRC (451 samples)
- LGG (513 samples)
- LUAD (542 samples)
- OV (469 samples)
- READ (122 samples)
- SKCM (366 samples)
- To encapsulate these rawMAFs we've added 2 new Archive Types:
- Mutation_Packager_Raw_Calls: rawMAFs
- Mutation_Packager_Raw_Coverage: rawWIGs
- New RPPA:
- KICH (63 samples)
- MESO (63 samples)
- BLCA, LGG, KIRP, PAAD, and STAD also had new RPPA samples, but merging failed. See issues below.
- Updated clinical data stream (via the Clinical_Pick_Tier1 pipeline):
- SARC histological type now available
- Corrected issue where radiations.radiation.regimenindication was set to "no" if patient.radiations.radiation.regimenindication contained "adjuvant". It is now set to "yes" in such a case.
- 185 mRNASeq RSEM aliquots (TP=184,TM=1) added to ESCA cohort (esophageal), and ergo STES as well (aggregate STAD+ESCA cohort).
- Issues:
- COAD cohort missing RPKM aliquots: due to a preprocessor failure the RPKM mRNASeq aliquots for the following patients
TCGA-AA-3841, TCGA-AA-3866,TCGA-AA-3870, TCGA-AA-3939, TCGA-AA-3941,TCGA-AA-3979, TCGA-AA-3980, TCGA-AA-3970,TCGA-AA-3977, TCGA-AA-3968are missing from our COAD stddata archives and FireBrowse. RSEM aliquots for these patients ARE available, however, and in general RSEM is preferred over RPKM for analysis. Finally, RPKM aliquots for these patients can be found in the aggregate COADREAD archives and FireBrowse database/api.
- RPPA failures due to changes in new data file row names: these have been reported to MDACC, but cannot be included in our stddata run until a comprehensive solution is available
- BLCA: no source/validation suffixes (batch 1.1.0)
- KIRP: ARHI-M-E -> DIRAS3-M-E (batch 2.0.0)
- LGG: Acetyl-a-Tubulin-Lys40-R-C -> Acetyl-a-Tubulin(Lys40)-R-C (batch 2.0.0)
- PAAD: ARHI-M-E -> DIRAS3-M-E (batch 2.0.0)
- PRAD: ADAR1-R-V -> ADAR1-M-V (batch 2.0.0)
- STAD: ADAR1-R-V -> ADAR1-M-V (batch 2.0.0)
- COAD cohort missing RPKM aliquots: due to a preprocessor failure the RPKM mRNASeq aliquots for the following patients
Summary of sample changes (see the comprehensive samples report for more details):
Clinical +252 (10945 total) MAF +125 (6584 total) miRSeq +1 (10160 total) RPPA +188 (5456 total) - New/Updated MAFs:
- CHOL (35 Samples)
- PAAD (+23 Samples)
- STAD (+68 Samples)
- TGCT (-1 Sample)
- Added new aggregate cohort STES, combining Stomach adenocarcinoma (STAD) and Esophageal carcinoma (ESCA) into a 628-patient cohort
- Updates to FireBrowse portal:
- Loaded with this stddata__2015_04_02 run
- Rationalized "requiredness" to be consistent across entire API:
- Now, nearly all functions require that at least one gene OR cohort OR barcode be specified
- This increases query performance, and clarifies API semantics
- New Quartiles API for mRNASeq expression, providing a summary of expression across all samples in a disease cohort, suitable e.g. for boxplot visualization
- Corrected docs for SampleTypeBarcode function to indicate that it takes only 1 barcode (as a "path parameter")
- Changes to the default sort parameter for several API functions:
- mRNASeq api and miRSeq functions now default to disease cohort instead of gene and mir, respectively
- significantly mutated genes and mutation file functions now default to disease cohort instead of Q value and gene, respectively
- When iterating over multiple pages of TSV, CSV output, the api will now return a header for ONLY the first page
- Columns containing commas are now delimited with double quotes in CSV return payloads
- Increased tolerance for messy input: distinct values of multi-valued parameters (e.g. gene=egfr,tp53,BRCA1 in mRNASeq samples api) may now be delimited by arbitrary whitespace (in addition to commas, as previously supported)
- In the interactive docs UI the
pageandpage_sizeparameters are no longer required by any API function: they continue to default to reasonable values for the common case. - The payload of the SampleTypes api now returns the description of the sample type (e.g. TP = tumor primary)
An empty response is now returned when no records in FireBrowse match a given query; previously a payload such as
{
"mRNASeq": []
}would be returned in such cases (e.g. for this mRNASeq query).
- removed spurous tcga_aliquot_barcode column from payload of miRSeq samples function (was visible in TSV and CSV return formats)
- avoid MongoDB conflict (and exception thrown) with certain MetaData/Counts api calls (reported by M. Deng)
- ISSUES: In the prostate (PRAD) RPPA data, the antibody source for ADAR1 changed from Rat to Mouse (ADAR1-R-V → ADAR1-M-V) between 1.0.0 and 2.0.0; this broke the Firehose sample merge pipelines, which requires all row names for each sample to match in order to generate a consistent table samples table.
Summary of sample changes (see the comprehensive samples report for more details):
BCR
-1
(11352 total)
Clinical
+551
(10693 total)
CN
-1
(10987 total)
MAF
+244
(6459 total)
Methylation
+532
(10955 total)
miRSeq
+10
(10159 total)
mRNASeq
+18
(10095 total)
RPPA
+270
(5268 total)
- New/Updated MAFs:
TGCT (testicular germ cell tumors) 150 samples
HNSC (head and neck squamous cell carcinoma) 279 samples (-27)
LGG (lower grade glioma) 286 samples (-3)
PAAD (pancreatic adenocarcinoma) 123 samples (+32)
PRAD (prostate adenocarcinoma) 425 samples (+92)
- New aggregate cohort: KIPAN
- Pan-kidney cohort of 973 patient cases, combined from KICH, KIRC, KIRP
- FireBrowseupdates:
- Loaded this 2015_02_04 data run
Expose the Link header within the response header payload of each API request, for CORS-enabled pagination within Javascript codes.
- By default only RSEM expression values will be returned now for mRNASeq api calls, instead of a mix of RSEM and RPKM values (which could be confusing). RPKM expression values can still be obtained by including a
&protocol=RPKMparameter.
Summary of sample changes (see the comprehensive samples report for more details):
BCR +39 (11353 total) Clinical +549 (10142 total) CN +2 (10988 total) MAF +154 (6215 total) Methylation +1 (10423 total) miRSeq +98 (10149 total) mRNASeq +347 (10077 total) RPPA +662 (4998 total) - Two new cohorts:
- GBMLGG: 1129 cases from GBM and LGG cohorts, combined into a single Glioma cohort for aggregate analysis
Note that due to the use of different antibodies for GBM & LGG we are unable to merge the RPPA data into a single table for this cohort - FPPP : FFPE Pilot Phase II
- GBMLGG: 1129 cases from GBM and LGG cohorts, combined into a single Glioma cohort for aggregate analysis
- FireBrowse:
- Loaded with this latest data run
- Exposed tool parameter in Archives/StandardData api call
- Run scheduling: given slowdown of data flow in TCGA, coupled with staff reductions, we are again adjusting our run schedule:
- Standard data runs will now be initiated bi-monthly (~8 weeks apart) instead of every 6 weeks
- Standard analysis runs will still be performed for every other data run, but that will result in 3 a year now instead of 4
- Custom AWG runs will still be performed on an as-needed basis
Summary of sample changes (see the comprehensive samples report for more details):
Clinical
+458
(9593 total)
CN
+1152
(10986 total)
MAF
-12
(6061 total)
miRSeq
+1031
(10051 total)
mRNASeq
+829
(9730 total)
- FireBrowse Updates:
- Updated to reflect 2014_10_17
- Added beta Analyses/FeatureTables api
- Properly terminate final row of returned TSV and CSV files, with newline (\n)
Summary of sample changes (see the comprehensive samples report for more details):
Clinical
+441
(9135 total)
CN
+360
(9834 total)
MAF
+155
(6073 total)
Methylation
+513
(10422 total)
miRSeq
+145
(9020 total)
mRNASeq
+353
(8901 total)
RPPA
+303
(4336 total)
- mRNASeq data:
- Z-score now computed for RSEM/RPKM mRNAseq, calculated as
z = (expression in tumor sample - mean expression in all tumor samples) / standard deviation of expression in all tumor samples
- Z-score now computed for RSEM/RPKM mRNAseq, calculated as
- mirSeq data:
- miRseq_Mature_Preprocess pipeline updated to miRbase v21, will now generate same mature miR calls as BCGSC
- Clinical data updates:
- corrected value of dccuploaddate CDE to be an actual date value instead of text showing how the date would be computed
- normalized CDEs to contain underscores for readability: e.g. dccuploaddate became dcc_upload_date, etc
- this is evident in Merge_Clinical and Clinical_Pick_Tier1 pipelines
- as well as in the results returned by the FireBrowse clinical samples API
- this is evident in Merge_Clinical and Clinical_Pick_Tier1 pipelines
- 2 formerly PHI clinical data elements, race and ethnicity are now available in Clinical_Pick_Tier1 pipeline output
- FireBrowse Updates:
- Samples/mRNASeq API:
- new Z-scores (see above) added to results
- geneID now reflected in results, because in some cases the gene name is absent
- Samples/clinical samples API:
- tier1_cde_name parameter added, to allow pinpoint retrieval of specific CDEs
- date parameter removed, for consistency with other Samples APIs (only latest available data is served)
- Added Implementation Notes documentation at top of clinical API section
- New Metatdata/ClinicalTier1 API: lists names of all tier 1 clinical data elements (CDEs), unioned over all disease cohorts
- New comprehensive file download dialogue, per disease cohort:
- See for example this example for adrenocortical carcinoma (ACC)
- This enables one to see all files for any disease cohort, in a single view
- And will be enhanced in the near future to have checkboxes to include/exclude files for aggregate download
- Also linked to the data column for each disease cohort on main GDAC site
- Samples/mRNASeq API:
Summary of sample changes (see the comprehensive samples report for more details):
BCR
+418
(11314 total)
Clinical
+195
(8694 total)
CN
+128
(9474 total)
MAF
+178
(5918 total)
Methylation
+262
(9909 total)
miRSeq
+73
(8875 total)
mRNASeq
+193
(8548 total)
RPPA
+1
(4033 total)
- Clinical data:
- Race and ethnicity parameters will now appear in Clinical_Pick_Tier1 output, whenever available
- Updates to dashboard on home page:
- stddata__YYYY_MM_DD version added to popup which appears when one clicks on Cases column
- TCGA data usage policy pops up first time one clicks on a Download link
- Update to FAQ:
- added new section for the TCGA Annotation Manager
- Improved navigability by adding a table of contents
Summary of sample changes (see the comprehensive samples report for more details):
BCR
+365
(10896 total)
Clinical
+191
(8499 total)
CN
+180
(9346 total)
Methylation
+272
(9647 total)
miRSeq
+601
(8802 total)
mRNASeq
+76
(8355 total)
- Switched LIHC MAF to the one produced by Baylor, as they are the GSC of record
Summary of sample changes (see the comprehensive samples report for more details):
BCR
+360
(10531 total)
Clinical
+218
(8308 total)
MAF
+202
(5740 total)
Methylation
+147
(9375 total)
mRNASeq
+255
(8279 total)
- New MAF for LIHC (Liver hepatocellular carcinoma, see our MAF dashboard for more details)
Summary of sample changes (see the comprehensive samples report for more details):
BCR +185 (10171 total) Clinical +170 (8090 total) CN +219 (9166 total) LowP -1 (1091 total) Methylation +263 (9228 total) miRSeq +208 (8201 total) mRNASeq +131 (8024 total) RPPA -1 (4032 total) - New MAFs (see our MAF dashboard for more details):
- ACC
- KIRP
- Changes to data preprocessing modules:
- All preprocessor modules are now executed as part of the standard data runs
- Previously only mRNAseq_Preprocess was executed during our stddata workflow
- And all other preprocessors were executed as preliminary tasks in our Analysis workflow
- The following modules have therefore been migrated from the analysis workflow to stddata workflow:
- Methylation_Preprocess
- mRNA_Preprocess_Median
- miRseq_Preprocess
- miRseq_Mature_Preprocess
- In addition, the code for the folllowing preprocessors has been updated:
- miRseq_Mature_Preprocess:
- corrected the output file name from "PKM" to "RPM"
- An additional output file created, of matrix without log2 transform
- mRNAseq_Preprocess: addtional output file created, of matrix without log2 transform
- miRseq_Mature_Preprocess:
- Merge_Clinical module updated: output will now include biospecimen information (BCR), when available
Summary of sample changes (see the comprehensive samples report for more details):
BCR
+271
(9986 total)
Clinical
+140
(7920 total)
CN
+176
(8947 total)
MAF
+147
(5538 total)
Methylation
+376
(8965 total)
miRSeq
+523
(7993 total)
mRNASeq
+260
(7893 total)
- New MAFs: see our MAF dashboard for more details
- BLCA (Bladder Urothelial Carcinoma)
- PAAD (Pancreatic adenocarcinoma)
- PRAD (Prostate adenocarcinoma)
- UCS (Uterine Carcinosarcoma)
- As foreshadowed in our February 2014 release notes, as of this run PANCAN datasets are no longer included
Summary of sample changes (see the comprehensive samples report for more details):
BCR
+232
(9715 total)
Clinical
+187
(7780 total)
CN
+284
(8771 total)
LowP
+1
(1092 total)
MAF
+56
(5391 total)
Methylation
+75
(8589 total)
mRNASeq
+155
(7633 total)
- New MAFs: see our MAF dashboard for more details
BRCA
- KIRP
- We intend to discontinue PANCANxx datasets in March 2014, unless we hear a strong desire from the community that they be continued.
- Clinical data for Adrenocortical Carcinoma (ACC) have been restored. See 2014_02_13 redaction notice below for more details.
Due to a PHI breach at one of our upstream data sources, where protected health information was accidentally included in the clinical data files for the Adrenocortical Carcinoma disease study (ACC), we have deleted all of the ACC stddata archives for the following tasks
- Clinical_Pick_Tier1
- Merge_Clinical
Summary of sample changes (see the comprehensive samples report for more details):
BCR
+318
(9483 total)
Clinical
+84
(7593 total)
CN
+210
(8487 total)
LowP
+53
(1091 total)
MAF
+329
(5335 total)
miRSeq
+79
(7470 total)
mRNASeq
+128
(7478 total)
- Samples Report now loads much faster: bulky sections (Filtered Samples, FFPE Cases, and Additional Annotations) converted to links
- mRNASeq Preprocessor updated to merge Illumina GA2 and Illumina HiSeq data if both are available in similar formats (either RPKM or RSEM)
- Our previous approach (of not merging) was unnecessarily conservative, because samples are typically NOT sequenced on both platforms
- For example: patients A, B sequenced only on Illumina GA2, and patients Y, Z sequenced only on Illumina HiSeq
- Previously, Illumina HiSeq data was used exclusively in downstream analyses when available (RSEM preferred over RPKM).
- This merge should be evident as significantly higher sample counts in downstream mRNASeq analyses (and reports)
- New / Updated MAFS:
- LGG
- UCS
- BRCA
- New MESO disease type (mesothelioma)
Summary of sample changes (see the comprehensive samples report for more details):
BCR
+221
(9165 total)
Clinical
+129
(7509 total)
CN
+57
(8277 total)
LowP
+179
(1038 total)
MAF
+313
(5006 total)
Methylation
+469
(8514 total)
miRSeq
+107
(7391 total)
mRNASeq
+93
(7350 total)
- New/Updated MAFs:
- ACC (90 samples)
- LAML (55 new samples)
- PRAD (168 new samples)
- New archives were uploaded by UNC to address GBM miRNAseq issue reported last month (2013_11_14 stddata run)
- Part 1 of 2 major upgrades to the Broad GDAC website: be sure to flush your cache to see the new layout
- Part 2 will be available with the next analysis run release, converging the stddata and analysis dashboards into a single, integrated entity
Summary of sample changes (see the comprehensive samples report for more details):
BCR
+199
(8944 total)
Clinical
+17
(7380 total)
CN
+66
(8224 total)
MAF
-55
(4693 total)
Methylation
+27
(8053 total)
miR
+74
(1135 total)
miRseq
+49
(7292 total)
mRNA
-2
(2217 total)
mRNAseq
+366
(7257 total)
- GBM: issue with miRNA Data
- Probe names in
unc.edu_GBM.H-miRNA_8x15K.Level_3.1.9.0did not match those of previous batches. UNC is looking into this. - No new miRNA merger in this stddata run.
- Probe names in
- Clinical_Pick_Tier1 pipeline: the following clinical features have been added:
- patient.clinicalcqcf.gleasonscorecombined (gleason_score_combined)
- patient.clinicalcqcf.gleasonscoreprimary (gleason_score_primary)
- patient.clinicalcqcf.gleasonscoresecondary (gleason_score_secondary)
- patient.clinicalcqcf.highestgleasonscore
- patient.stageevent.gleasongrading.gleasonscore
- patient.clinicalcqcf.psaresultpreop (psa_result_preop)
- patient.clinicalcqcf.daystopreoppsa
- patient.stageevent.psa.psavalue
- patient.stageevent.psa.daystopsa
Corrected several broken links referring to "runs/latest_analyses" on the FAQ and Analysis Run release notes pages
Summary of sample changes (see the comprehensive samples report for more details):
BCR
+197
(8745 total)
Clinical
+28
(7363 total)
CN
+192
(8158 total)
MAF
+80
(4748 total)
Methylation
+1
(8026 total)
miRseq
+79
(7243 total)
mRNAseq
+1
(6891 total)
RPPA
+1
(4033 total)
- Merge_Clinical updated to remove duplicates in sample IDs of clinical data
- As mentioned in last month's stddata run, FFPE aliquots are now segregated into distinctly-named <Disease_Cohort>-FFPE archives
The snapshot was taken early this month (2013_10_10, instead of 2013_10_15) due to The Broad Institute IT Outage
Summary of sample changes (see the comprehensive samples report for more details):
BCR
-1
(8548 total)
Clinical
+176
(7335 total)
CN
+97
(7966 total)
LowP
-5
(859 total)
MAF
-1
(4668 total)
Methylation
+142
(8025 total)
miRseq
+274
(7164 total)
mRNAseq
+549
(6890 total)
RPPA
+685
(4032 total)
- Please note that we've introduced a new procedure for handling FFPE aliquots in our stddata processing: even though it has been shown that with sufficient care it is possible to perform TCGA-like cancer genome analyses on FFPE samples, confounding batch effects may be introduced if FFPE aliquots are combined with frozen tissue aliquots in the same analysis. To avoid these and maintain the highest quality downstream analyses, as of this September 2013 run we are segregating FFPE samples from frozen tissue samples; this is reflected as a small decrease in the sample counts of our uploaded stddata archives. If you wish to perform analyses on FFPE-only sample sets, they will appear in the October 2013 stddata run as distinctly-named <Disease_Cohort>-FFPE archives.
- Increased number of platforms/datasets for Adrenocortical carcinoma (ACC) and Uterine Carcinosarcoma (UCS) from 1 and 8 to 14 and 16, respectively.
- Released v0.4.2 of firehose_get
Summary of sample changes (see the comprehensive samples report for more details):
BCR
+131
(8549 total)
Clinical
+149
(7159 total)
CN
+469
(7869 total)
LowP
-6
(864 total)
MAF
+61
(4669 total)
Methylation
+185
(7883 total)
miRseq
-5
(6890 total)
mRNAseq
+183
(6341 total)
Updated pipeline: Clinical_Pick_Tier1: the clinical parameter selection file is now available in stddata run package. This file defines what clinical features are selected for analysis run and how their values are transformed for each disease type.
- Due to an issue with the UUID mapping service at the DCC, we based this August 2013 run on a slightly earlier (than 8/15) snapshot of data. The issue has been reported to DCC and should not affect the next data run (in September).
Summary of sample changes (see the comprehensive samples report for more details):
BCR
+45
(8418 total)
Clinical
+102
(7010 total)
CN
-2
(7400 total)
Methylation
+55
(7698 total)
miRseq
+200
(6895 total)
mRNAseq
-2
(6158 total)
RPPA
+41
(3347 total)
- There was an issue with the RPPA data for LUAD: two rows for Bcl-2, each with different values. M. D. Anderson has been notified, and it is being corrected. Please use last month's RPPA data if you are analyzing LUAD.
- Reflects 3 pipelines for new UCS disease cohort (Uterine Carcinosarcoma)
- Clarified how followup data are systematized during our clinical data processing: that is, for each patient we
- Check vital status
For each patient,- vistal status
1. if any version of vital status parameter value is 'deceased' then fill the picker vital status parameter value with 'deceased'
2. if not, fill the picker vital status parameter value with 'living'
=> in selectionFileGenerator.R in picker module
followup.vitalstatus <- paste("=ifelse(",paste(rownames(patientData)[w.vitalstatus], "==\"deceased\"",collapse=" | ") ,",1, 0)") - days to last followup1. First check vital status,if the patient's vital status parameter value is 'deceased', then fill the 'days to last followup' parameter value with 'NA'.
2. if the patient's vital status parameter value is 'living' then, go to parameter 'days to last followup' and check all versions of'days to last follow up' parameters and pick up the maximum number of the parameter values.
=> in selectionFileGenerator.R in picker module
followup.daystolastfollowup <- paste("ifelse(", any.deceased, ",\"NA\",", max.any.daystolastfollowup,")", sep="") - days to death
1. if the values of all version of days to death parameter is NA then fill the days to death parameter value with NA
2. if not, fill the days to death parameter value with the maximum number of the parameter values among any version of days to death parameters values
=> in selectionFileGenerator.R in picker module
followup.daystodeath <- paste("ifelse(", all.na,",\"NA\", max(", any.daystodeath, ",na.rm=T))", sep="")
- vistal status
- Check vital status
- IMPORTANT: this will be the final installment of our bi-monthly cycle of Firehose stddata package releases. In order to better focus on serving AWG needs, evolving computational methods, and the portals used to disseminate & explore them, we are switching to a monthly release cycle. Our next Firehose stddata run will be cut from data snapshot in mid July 2013, and released shortly thereafter.
Summary of sample changes (see the comprehensive samples report for more details):
BCR
+110
(8373 total)
Clinical
+166
(6908 total)
MAF
+123
(4608 total)
Methylation
+379
(7643 total)
miRseq
+375
(6695 total)
mRNAseq
+279
(6160 total)
RPPA
+73
(3306 total)
New MAF files (see MAF Dashboard for more details):
- THCA
- KIRC
Summary of sample changes (see the comprehensive samples report for more details):
BCR
-4
(8263 total)
Clinical
+211
(6742 total)
CN
-1
(7402 total)
LowP
-1
(870 total)
MAF
+80
(4485 total)
Methylation
-2
(7264 total)
miRseq
-2
(6320 total)
mRNAseq
+63
(5881 total)
RPPA
-2
(3233 total)
- New THCA MAF file (see MAF Dashboard for more details)
- Everything uploaded to DCC, available in IGV and downloadable via firehose_get (which has been updated to v0.3.13, with updated online docs & examples)
Summary of sample changes (see the comprehensive samples report for more details):
BCR
+236
(8267 total)
Clinical
+34
(6531 total)
CN
+111
(7403 total)
LowP
+120
(871 total)
MAF
+105
(4405 total)
Methylation
+119
(7266 total)
miRseq
+1
(6322 total)
- New AWG-curated MAF for STAD (see MAF Dashboard for more details)
- Everything uploaded to DCC, available in IGV and downloadable via firehose_get
(previous bandwidth problems were determined not to be issues internal to the Broad, and were reported to external stakeholders) - BCR data for ACC (adrenocortical carcinoma) and UCS (uterine carcinosarcoma) disease studies have appeared in our stream, but nothing else is available for external packaging
Summary of sample changes (see the comprehensive samples report for more details):
BCR
+188
(8031 total)
Clinical
+99
(6497 total)
CN
+97
(7292 total)
LowP
+115
(751 total)
MAF
+11
(4300 total)
Methylation
+1
(7147 total)
miRseq
+101
(6321 total)
mRNAseq
+162
(5818 total)
- LowPass data now included in IGV release
PAAD reintroduced, without uncertified samples removed last month
- Mutation Data (for more details see our MAF dashboard):
- New AWG-curated MAF for SKCM
- Updated MAF for LAML
- These data are available via firehose_get and IGV; upload to the DCC is in process, but incomplete due to continuing bandwidth issues; we will notify when the data are fully reflected at the DCC
Sample Changes: for more details please consult our samples summary report
BCR
-73
(7843 total)
Clinical
+154
(6398 total)
CN
-30
(7195 total)
MAF
-34
(4289 total)
Methylation
-49
(7146 total)
miRseq
+101
(6220 total)
mRNAseq
+276
(5656 total)
RPPA
+62
(3235 total)
- All pancreatic adenocarcinoma (PAAD) data have been removed due to IRB issues; they will appear again in the first May 2013 stddata run
- As of 2013_05_08 these data are available via firehose_get and IGV; upload to the DCC is in process, but incomplete due to a low bandwidth issue; we will notify when the data are fully reflected at the DCC.
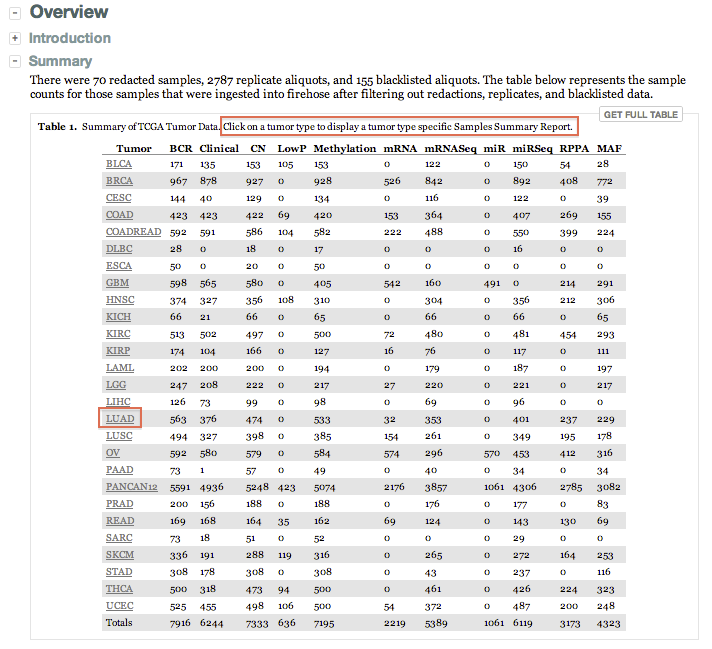
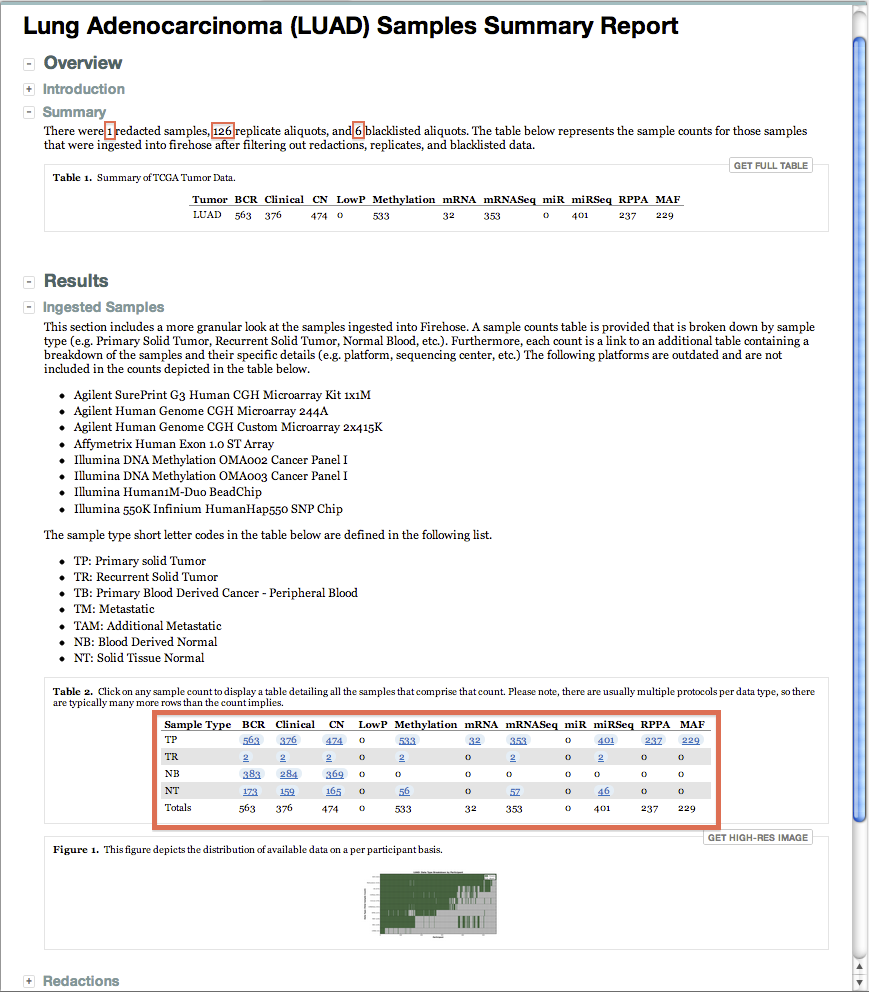
- The Samples Summary Report for each run now reflects significantly higher-resolution metadata per disease cohort:
- In addition to the combined list of all excluded samples (across all diseases) the report now also lists those per disease cohort
- Consider e.g. the LUAD example below: 1 sample was redacted, 126 were filtered as replicates, and 6 were blacklisted
- Data resolution down to the aliquot is now reflected in the report, for samples both included and excluded
- The sequencing platform, submitting center, data level, and protocol are also reflected
- As well as the sample type (e.g. primary tumor, recurrent tumor, normal blood, etc.) for each ingested aliquot
- We believe this level of clarity, comprehensiveness, and ease of access raises the bar for data resolution in TCGA
- Blacklisted 138 STAD copy number samples (from batch 220, determined by A. Cherniack to have wrong sample names)
BCR | +116 | (7916 total) |
|---|---|---|
Clinical | +4 | (6244 total) |
MAF | +123 | (4323 total) |
mRNAseq | +52 | (5380 total) |
Disease | Tumors | Normals | Mutations |
|---|---|---|---|
KICH | 65 | 65 | 1788 |
KIRP | +11 (114) | +14 (123) | +1074 (10131) |
LGG | +47 (217) | +47 (217) | +2626 (27275) |
PAAD | +0 (34) | +0 (34) | -28 (6442) |
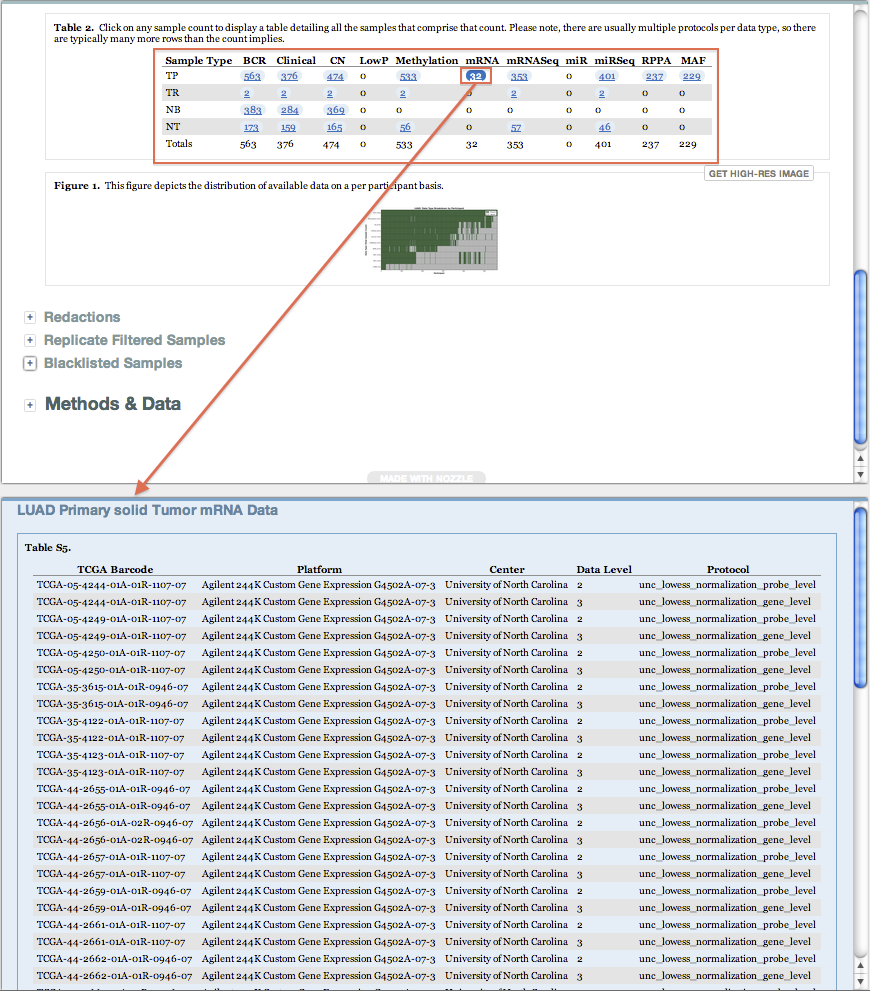
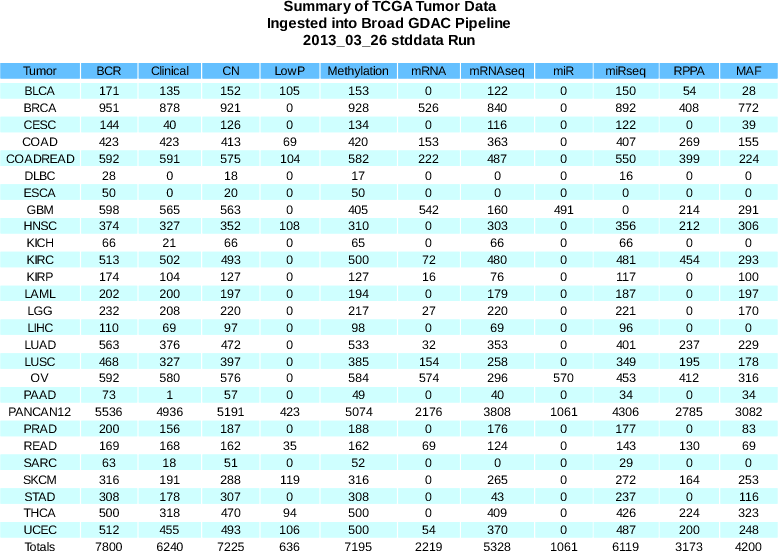
Sample Changes:
BCR
+21
(7800 total)
Clinical
+106
(6240 total)
CN
+206
(7225 total)
Methylation
+167
(7195 total)
miRseq
+60
(6119 total)
mRNAseq
+315
(5328 total)
- New UCEC MAF (+37 Mutations)
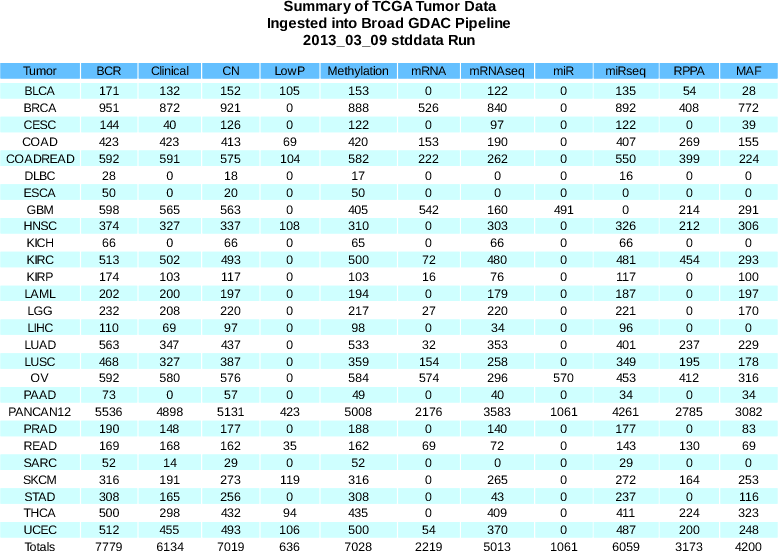
Sample Changes
BCR
+319
(7779 total)
Clinical
+27
(6134 total)
CN
+163
(7019 total)
Methylation
+269
(7028 total)
miRseq
-1
(6059 total)
mRNA
-3
(2219 total)
mRNAseq
-5
(5013 total)
- Presented detailed scheme of MAF processing on IWG Call:
- GBM MAF updated to sync with DCC
- Minor organizational & documentation enhancements to GDAC site and dashboards
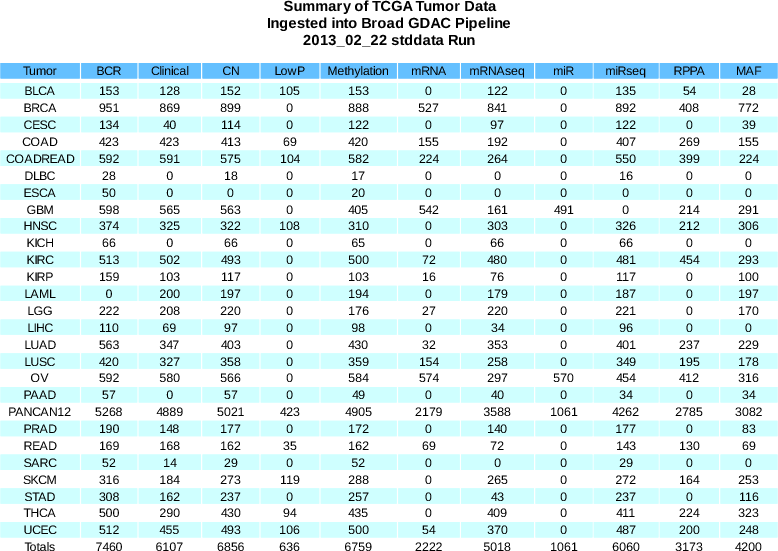
Sample Changes
BCR
-8
(7460 total)
Clinical
+68
(6107 total)
MAF
+266
(4200 total)
Methylation
+23
(6759 total)
miRseq
-2
(6060 total)
mRNAseq
+85
(5018 total)
- Clinical parameter update for THCA
- Additional 9 clinical parameters are included for THCA.
- 5 parameters for tumor aggressiveness (distant.metastasis, extrathyroidal.extension, lymph.node.metastasis, completeness.of.resection, number.of.lymph.nodes)
- 4 parameters for tumor staging systems for THCA (tumor.stagecode, neoplasm.diseasestage, multifocality, tumor.size)
- tumor.size = max(dimension.depth, dimension.length, dimension.width)
- MAFs updated to sync with DCC:
- BLCA v1.3
- BRCA v5.2 (+265 Tumor Samples)
- CESC v1.4 (+3 Tumor Samples)
- LAML v2.12 (-2 Tumor Samples)
- LUSC v1.5
- PRAD v1.3
- UCEC v1.4
- hg19 WIGs added:
- BRCA
- UCEC
- GBM
- Addressed UUID uppercase bug in our dicer which accidently removed 63 COAD RPPA samples; fix will be evident in data stream after SDRF is updated at DCC.
- Added Firehose AWG Runs page to the NCI Wiki, as a central reference point for custom runs done on behalf of active AWGs. This effort has several aims:
- Currency: Firehose can be run on the latest daily snapshot of data from the DCC, avoiding the time & sample lag of monthly runs
- Flexibility: additional runs can be easily performed on AWG-curated disease subtypes, and even include custom analyses
- Speed: custom AWG runs can be executed in only a few days time (excluding computationally intensive algorithms that may take >1 week to run)
- Familiarity: using the same internal Firehose machinery, and external-facing dashboards, Nozzle reports etc, already known to the community
- Scope: stepping stone to open-access Firehose, that can be manipulated directly by TCGA researchers instead of having runs curated only at the Broad
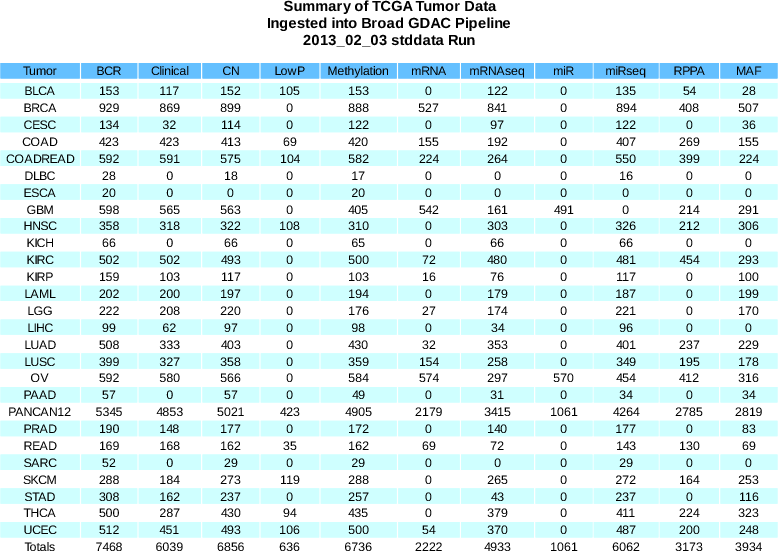
Sample count changes:
Clinical
+90
(6039 total)
CN
+304
(6856 total)
LowP
+35
(636 total)
Methylation
+118
(6736 total)
miRseq
+435
(6062 total)
mRNAseq
+143
(4933 total)
Methylation data newly available for ESCA
- These data are already reflected in IGV (File->Load From Server menu)
Note that firehose_get v0.3.10 was released on Jan 31, to dynamically reflect the range of disease cohort names used in runs
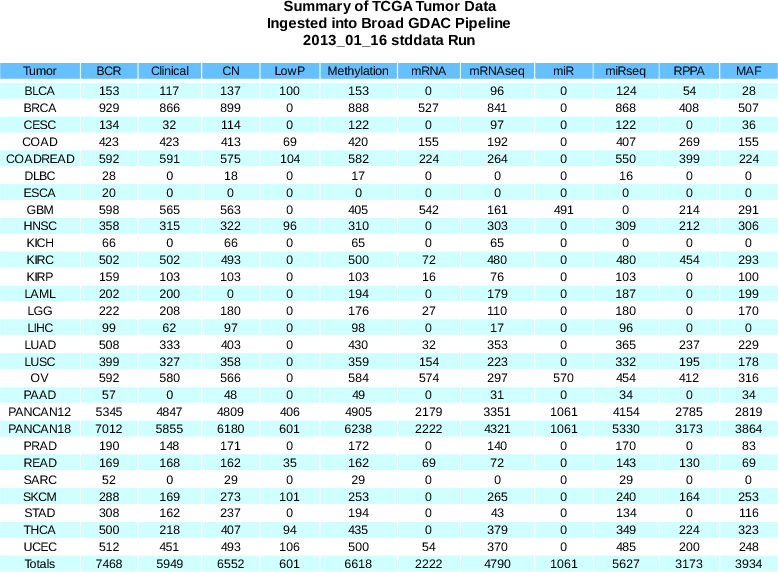
Sample Changes:
BCR
+161
(7468 total)
Clinical
+40
(5949 total)
CN
+122
(6552 total)
MAF
+136
(3934 total)
Methylation
+96
(6618 total)
mRNA
-2
(2222 total)
mRNAseq
+110
(4790 total)
- LGG MAF corrected (see 2012_12_21 errata below)
Given our desire to support TCGA AWGs more extensively in 2013, and that AWG runs are timed more to telecon & workshop schedules, this will be our only stddata run in January 2013. The next stddata run will be initiated in the first week of Feb, after our first complete Feb download from the DCC.
In a joint decision with the PANCAN awg, we will discontinue generating the PANCAN18 cohort until at least May 1 2013. For this run we have uploaded PANCAN18 to the DCC, but it will not be retrievable with firehose_get.
We have initiated the export to IGV of this run, and expect it to be completed by 10pm EST tonight and available to the public Sat Jan 26. Until then, your IGV menus will not show the 2013_01_16 run.
Due to a dicing issue, the PAAD MAF was processed rather than the LGG one for the 12/21 run. The correct file had been swapped in prior to the run, but the dicer did not recognize the swap, and didn't process the LGG MAF. Our next run will have the correct LGG MAF. This affects mutation results for LGG in the analyses__2012_12_21 run as well.
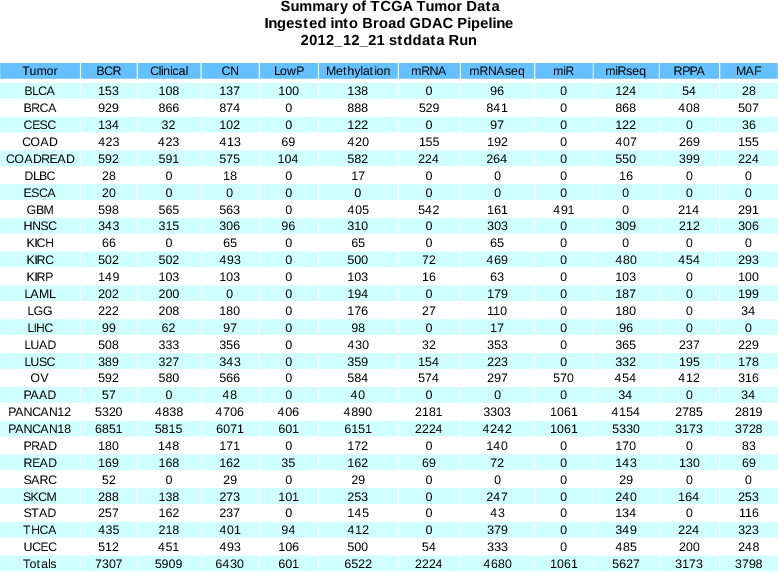
Sample Changes
BCR
+73
(7307 total)
CN
+12
(6430 total)
LowP
+100
(601 total)
MAF
+617
(3798 total)
mRNAseq
+323
(4680 total)
- Added New MAFs:
- SKCM (using latest version auto-submitted by GSC to DCC)
- KIRP (using latest version auto-submitted by GSC to DCC)
- LGG (using latest version auto-submitted by GSC to DCC)
- PAAD (using latest version auto-submitted by GSC to DCC)
- HNSC (using AWG manually-curated version)
please note that, per the AWG, this MAF contains contaminated samples; a new MAF is currently under construction by the AWG, and should be ready for inclusion in our next stddata run.
- Removed old v1.3 KIRC MAF (current version is 1.4)
- This accounts for the 110 sample drop for KIRC MAF tumor samples
- We are aware of a new v1.5 of the KIRC MAF posted to a non-standard location rather than the DCC. If appropriate, it will be added to our next stddata run, replacing v1.4 and bringing the KIRC MAF tumor sample count to 499.
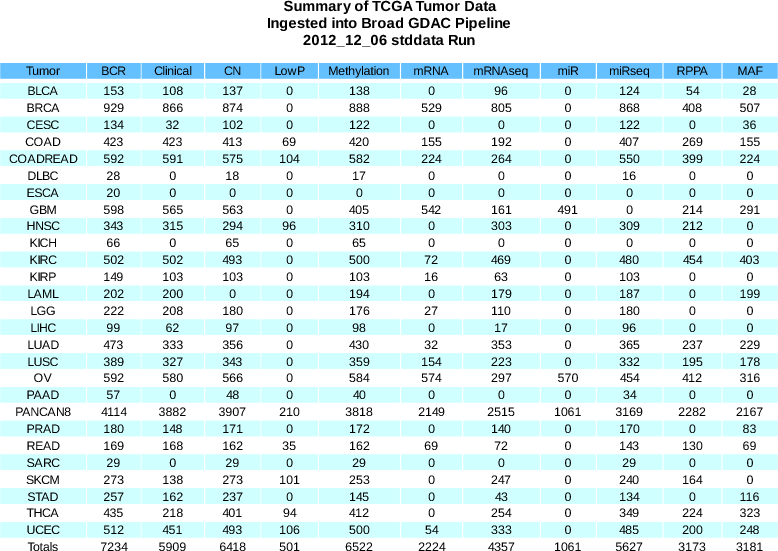
Sample Changes (Negative values reflect recent redactions - please see the Samples Summary Report)
BCR
+111
(7234 total)
Clinical
+111
(5909 total)
CN
+206
(6418 total)
MAF
-2
(3181 total)
Methylation
+58
(6522 total)
miRseq
-17
(5627 total)
mRNAseq
-14
(4357 total)
- New curated MAF from the GBM AWG - contains 291 sample (an increase of 15 samples).
- Level 3 Clinical Parameters updated:
- Addition of radiation exposure (corresponds to patient.personlifetimeriskradiationexposureindicator in Level 2 Data)
- Deletion of neoadjuvanttherapy (corresponded to patient.drugs.drug.regimenindication in Level 2 Data)
- Reflects the following Level 2 clinical parameters related to smoking to the extent available (for BLCA, CESC, HNSC, LUAD, LUSC, and PANCAN8 cohorts)
patient.numberpackyearssmoked
patient.stoppedsmokingyear
patient.tobaccosmokinghistoryindicator
patient.yearoftobaccosmokingonset
Our internal rewiring to accommodate normal samples induced a hiccup which required several MAF packager files to be reissued for the stddata__2012_10_24 run. The new files have been uploaded to the DCC and will be available in the protected data tree pending internal mirroring by the DCC, or may be downloaded presently via firehose_get. The newly issued files are identifiable as
*BRCA.Mutation_Packager_Calls.*.2012102400.1.0.*
*OV.Mutation_Packager_Calls.*.2012102400.1.0.*
*LAML.Mutation_Packager_Calls.*.2012102400.1.0.*
*UCEC.Mutation_Packager_Calls.*.2012102400.1.0.*
*PANCAN8.Mutation_Packager_Calls.*.2012102400.1.0.*
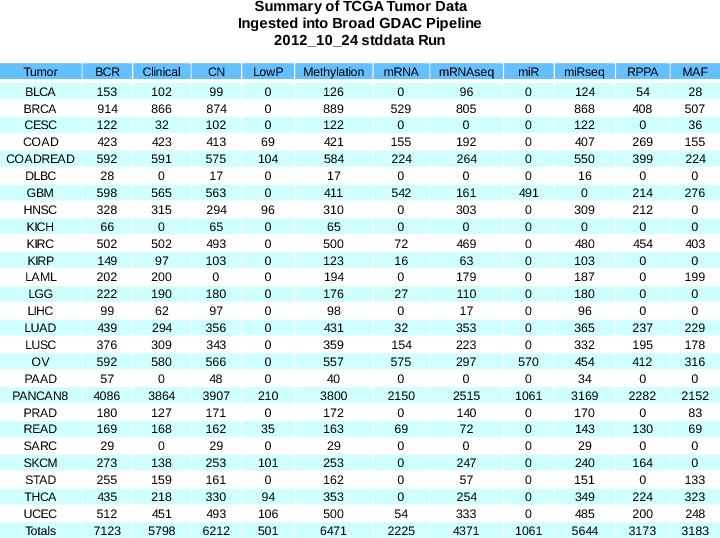
Tumor Sample Changes:
BCR
+38
(7123 total)
Clinical
+54
(5798 total)
CN
+34
(6212 total)
MAF
+323
(3183 total)
Methylation
+437
(6471 total)
miRseq
+851
(5644 total)
mRNA
+1
(2225 total)
mRNAseq
+547
(4371 total)
- Normal samples (where available) will henceforth be included for each platform:
- Yielding a total of 5544 archives submitted to the DCC (+2262 increase from the previous count of 3282)
- Two enhancements to samples summary report available on our dashboard:
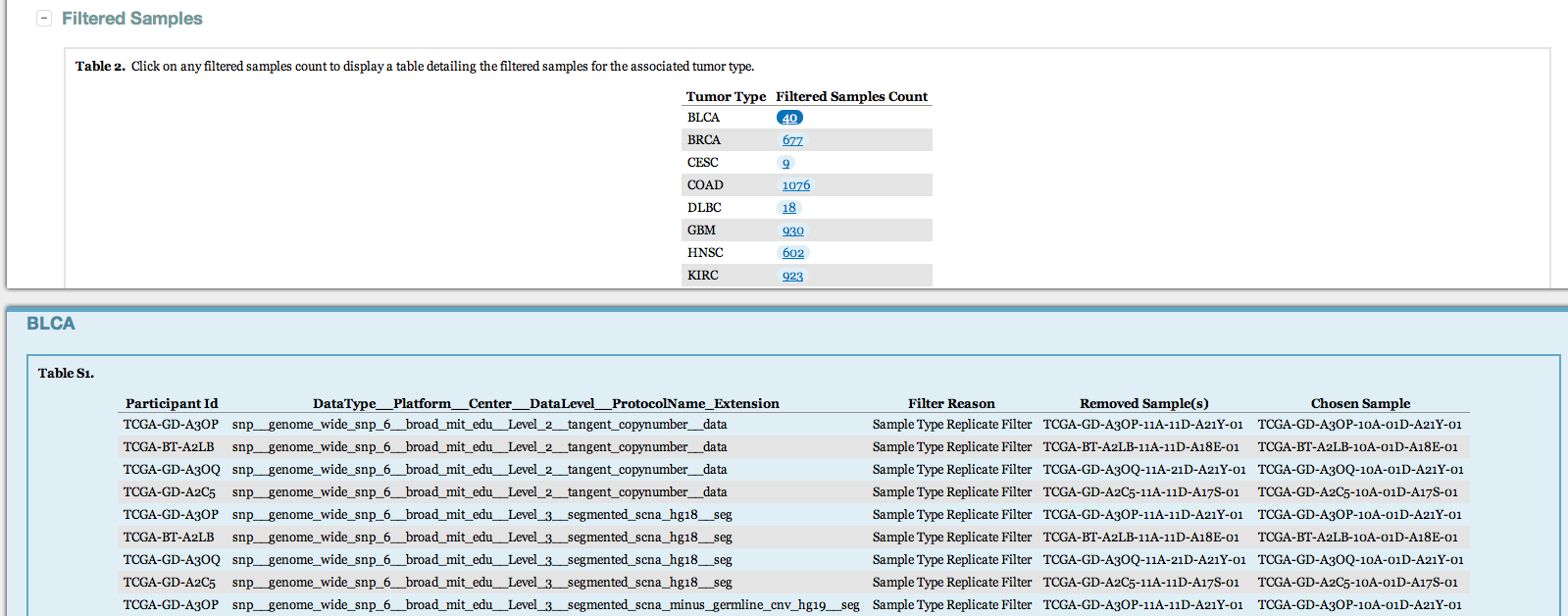
- Now lists every sample that is filtered from the datastream, with an explanation of why (as per our FAQ)
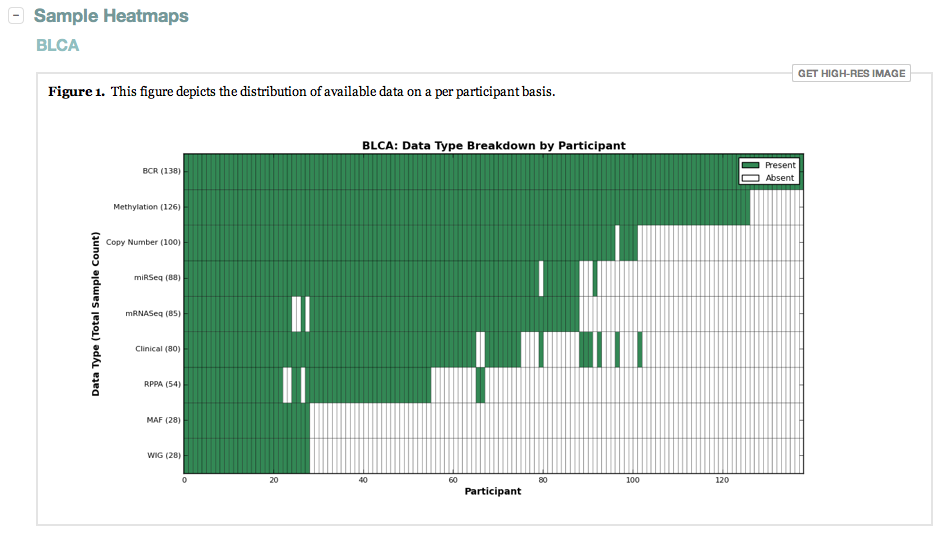
- Heatmaps are now included that display available samples per data type vs. participants
- Now lists every sample that is filtered from the datastream, with an explanation of why (as per our FAQ)
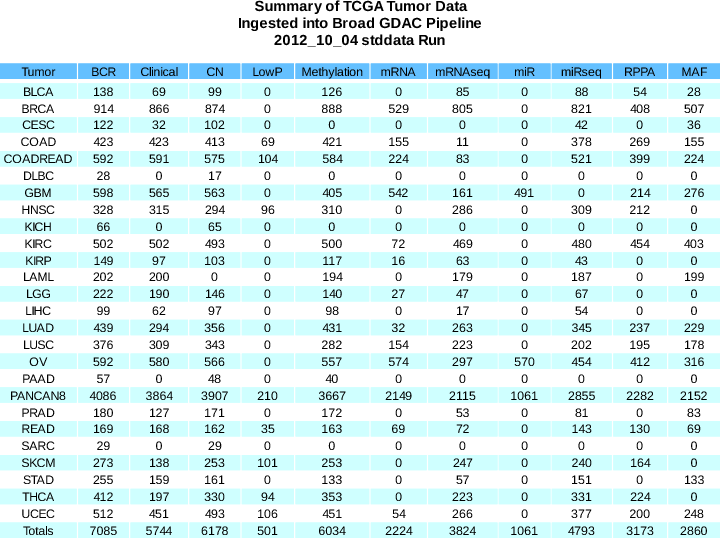
Sample Changes:
BCR
+133
(7085 total)
Clinical
+65
(5744 total)
CN
+364
(6178 total)
LowP
+23
(501 total)
Methylation
+445
(6034 total)
mRNAseq
+261
(3824 total)
- Methylation newly available for LIHC
- CN newly available for KICH
- mRNAseq newly available for GBM
- No significant sample changes 26 Aug - 06 Sept (see Spreadsheet).
To assist the melanoma AWG we delayed the first stddata run of Sept 2012 to incorporate pending submission of RPPA samples for SKCM, which appeared in our mirror as of 9/13. As this was essentially the midpoint of September
stddata__2012_09_13 WAS THE ONLY STDDATA RUN for SEPT 2012. This simplified our work somewhat, without appreciably reducing the sample flow, while also allowing us to re-sync to our desired target stddata run schedule of the 1st and 15th of each month.Sample Changes:
BCR
+71
(6952 total)
Clinical
+8
(5679 total)
CN
+4
(5814 total)
Methylation
+118
(5589 total)
miRseq
+684
(4793 total)
mRNAseq
+36
(3563 total)
RPPA
+442
(3173 total)
- The past 5 months of Standardized Data have been loaded into IGV:
- Partitioned by reference genome - When choosing "Load from Server...", only the data for the currently selected reference (Human hg18/Human hg19) will be available via the menu.
- Copy Number data available for both hg18 and hg19, both with and without germline samples
- Meth450 data now available
To access, open IGV, and with Human hg18/hg19 selected as the reference, navigate:
File -> Load from Server... -> The Cancer Genome Atlas -> TCGA GDAC -> Firehose Standard Data
- RPPA samples newly available for three tumor types:
- BLCA
- SKCM
- THCA
- Potential RPPA issues (waiting on confirmation from M.D. Anderson)
- Results may be reporting KDR rather than XIAP for KIRC and UCEC when converting using the supplied antibody annotations
- LKB1 antibody may be reported as LBK1 for BRCA and OV
- PANCANCER aggregate (containing 20+ disease types) replaced with PANCAN8 aggregate (containing only 8: BRCA COAD READ KIRC GBM LUSC OV UCEC)
- COAD and READ reintroduced as separate types by request of the PANCAN8 AWG (COADREAD aggregate continues to be available)
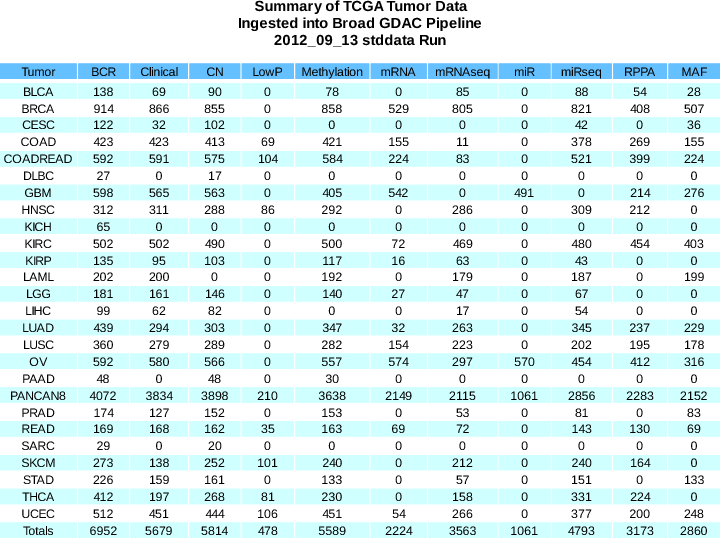
The LUAD mafs we ingested in stddata__2012_08_25 had non-standard headers. We've since enhanced our MAF ingestor to correct such headers when detected, and re-released the LUAD maf yesterday (17 Sept. 2012). New tarballs are available from firehose_get, and soon the DCC, as:
- gdac.broadinstitute.org_LUAD.Mutation_Packager_Calls.*.2012082500.1.0.tar.gz
- Dicer:
- Removed legacy internal sample blacklist (see Standardized Data Run Notes for 2012_08_04)
- Updated filtering
- File containing list of filtered samples: filteredSamples.2012_08_25__00_00_10.txt.
- Gene annotated protein expression data (RPPA) are now available for LUSC, LUAD, and HNSC.
- New SARC disease type reflected (Sarcoma)
Sample Changes
CN
+323
(5810 total)
Clinical
+31
(5671 total)
LowP
+402
(478 total)
Methylation
+7
(5471 total)
RPPA
+407
(2731 total)
mRNA
+7
(2224 total)
mRNAseq
+29
(3527 total)
miR
+7
(1061 total)
miRseq
+20
(4109 total)
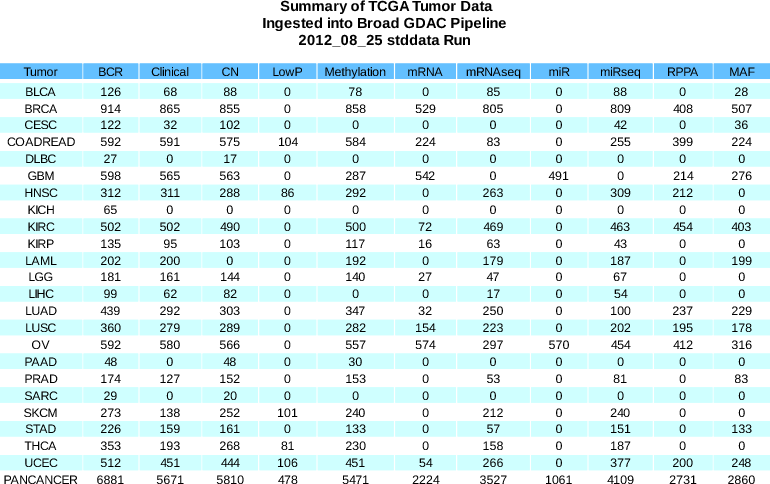
Sample Changes:
BCR
+35
(6881 total)
Clinical
+7
(5640 total)
CN
+101
(5487 total)
Methylation
-1
(5464 total)
miR
-1
(1054 total)
miRseq
+113
(4089 total)
mRNA
-1
(2217 total)
mRNAseq
+38
(3498 total)
RPPA
+237
(2324 total)
The -1's in this table were caused by the removal of several samples for two OV patients, due to an unintended clash between improved aliquot selection (as described in our FAQ) and a legacy internal blacklist from the TCGA pilot. The legacy blacklist has now been deprecated, in favor of relying solely on the transparency of redactions recorded in the DCC annotations database, and the samples will be present in future releases. The OV samples in question are:
- TCGA-29-1704
- CN
- TCGA-23-1023
- mRNA
- miR
- Methylation
- TCGA-29-1704
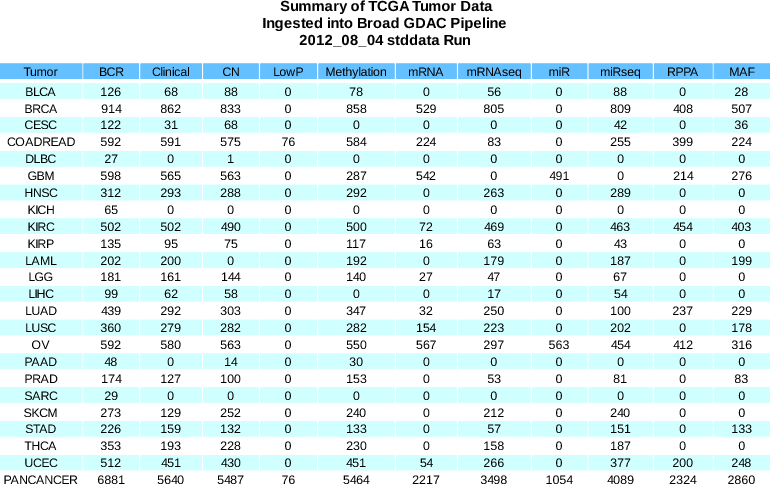
New Samples:
BCR +65 (6846 total) Clinical +6 (5633 total) Methylation +140 (5465 total) mRNAseq +170 (3460 total) miRseq +195 (3976 total) - Pipelines now recognize WashU as submitting BCR where applicable, which e.g. adds LAML clinical samples to our data stream.
- The WIGs we fabricate to simulate coverage for MutSig have been updated to hg19 for:
- BLCA
- BRCA
- CESC
- KIRC
- LUSC
- LUAD
- PRAD
- STAD
- UCEC
- 102 WIGs missing from COADREAD. Please see explanation in our email archive.
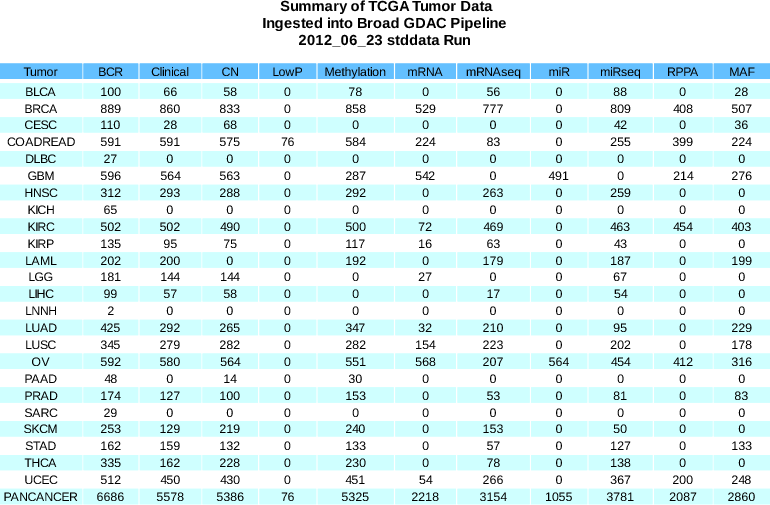
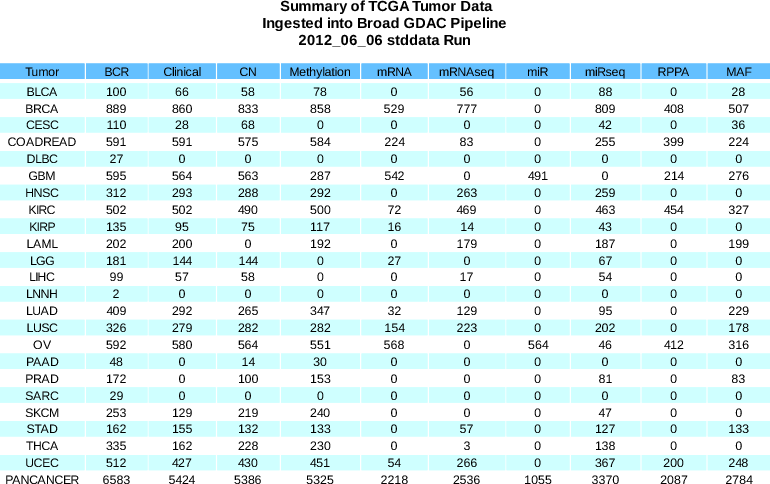
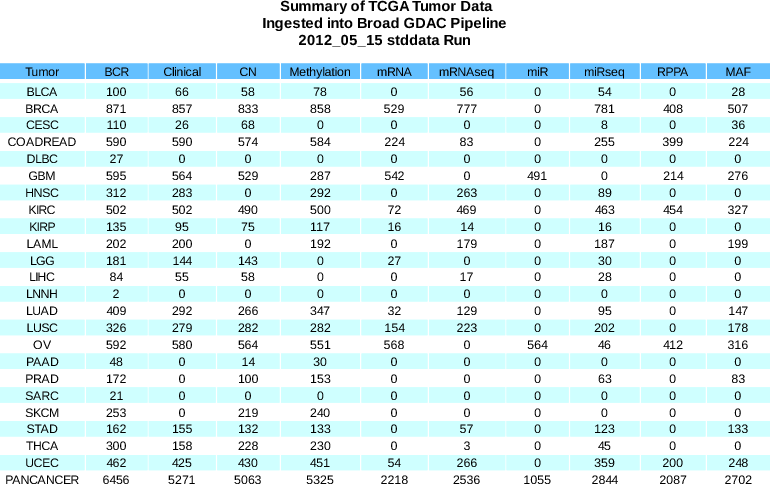
to address item (9) in our 2012_06_23 Analysis Run Release Notes
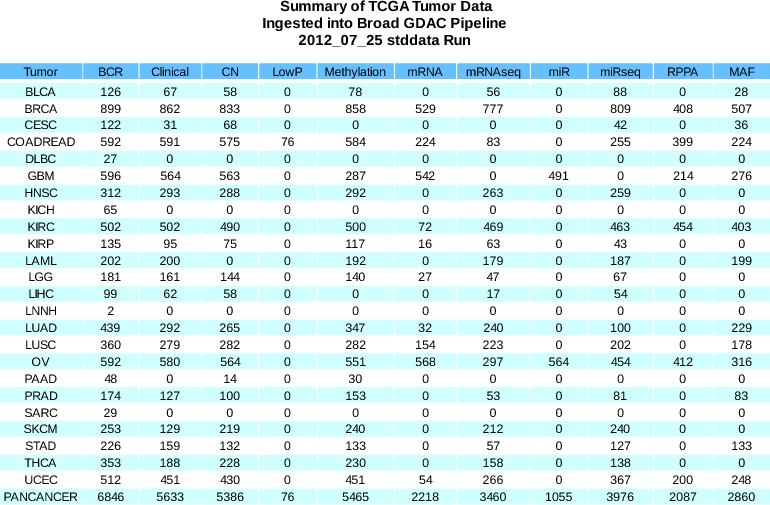
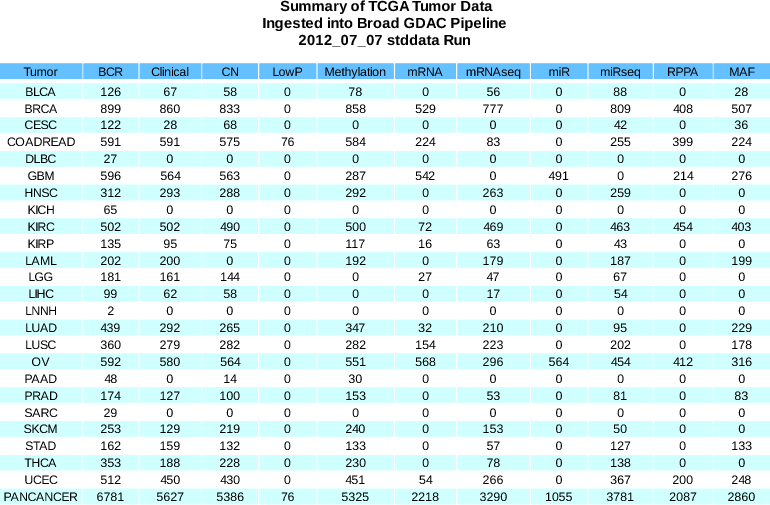
New Samples:BCR +95 (6781 total)Clinical +49 (5627 total)mRNAseq +136 (3290 total)
- New DNAseq column in data table, reflecting low-pass DNAseq copy number datatype from HMS:
Merge_cna__illuminahiseq_dnaseqc__hms_harvard_edu__Level_3__segmentation__seg
- New pipelines for RNAseq v2 data from UNC:
- Merge_rnaseqv2__illuminahiseq_rnaseqv2__unc_edu__Level_3__exon_quantification__data
- Merge_rnaseqv2__illuminahiseq_rnaseqv2__unc_edu__Level_3__junction_quantification__data
- Merge_rnaseqv2__illuminahiseq_rnaseqv2__unc_edu__Level_3__RSEM_genes_normalized__data
- Merge_rnaseqv2__illuminahiseq_rnaseqv2__unc_edu__Level_3__RSEM_genes__data
- Merge_rnaseqv2__illuminahiseq_rnaseqv2__unc_edu__Level_3__RSEM_isoforms_normalized__data
- New pipelines for HiSeq RNAseq data from BCGSC:
- Merge_rnaseq__illuminahiseq_rnaseq__bcgsc_ca__Level_3__exon_expression__data
- Merge_rnaseq__illuminahiseq_rnaseq__bcgsc_ca__Level_3__gene_expression__data
- Merge_rnaseq__illuminahiseq_rnaseq__bcgsc_ca__Level_3__splice_junction_expression__data
- 526 new miRseq samples and 153 new clinical samples
Mutation_Packager_Coveragetask:- Use soft links instead of packing multiple copies of identical coverage files, for dramatic disk savings: e.g. reduced archive size from 58Mb to 2 Mb for 28 BLCA samples, from 697Mb to 3 Mb for 248 UCEC samples, etc!
- Valid SDRF now included in DCC archive
Mutation_Packager_Calls task now includes valid SDRF in DCC archiveMerge_Clinical task now includes valid SDRF in DCC archive
- New RPPA_AnnotateWithGene pipeline, which:
- Maps antibody proteins to gene symbols, to simplify downstream analyses. The gene symbols come from the MDA_RPPA_Core.antibody_annotation.txt file packaged in the mage-tab archive of each RPPA submission.
- Includes a figure showing the protein expression levels per sample.
- Mutation_Packager: split into two files, Mutation_Packager_Calls and Mutation_Packager_Coverage, for smaller downloads if all one wants are MAFs.
- Prostate (PRAD) mutation data now present (83 samples)
- Per Program Office, all clinical and mutation data are now public, so we no longer cull them prior to hosting on Broad GDAC site.
New Broad GCC SNP6 data format, in which all samples from the single platform
have been reprocessed in 4 distinct ways (each containing the full complement of samples):
Merge_snp__genome_wide_snp_6__broad_mit_edu__Level_3__segmented_scna_hg18__seg
Merge_snp__genome_wide_snp_6__broad_mit_edu__Level_3__segmented_scna_hg19__seg
Merge_snp__genome_wide_snp_6__broad_mit_edu__Level_3__segmented_scna_minus_germline_cnv_hg18__seg
Merge_snp__genome_wide_snp_6__broad_mit_edu__Level_3__segmented_scna_minus_germline_cnv_hg19__seg
Please consult the Broad Institute GCC for details on how these data have changed.
Two BCGSC platforms have been manually deleted for LAML disease type:
mirnaseq__illuminaga_mirnaseq__bcgsc_ca__Level_3__isoform_expression__data
mirnaseq__illuminaga_mirnaseq__bcgsc_ca__Level_3__mirna_expression__data
Phase 2 SDRFs implementation: all Merge_<platform> pipelines now have SDRFs (clinical and MAF packages will be added next run)
- Redactions report now included on stddata dashboard
- Corrected 2 clinical issues: reducing to 5 the # patients with days_to_death=0, and ensuring vitalstatus=living implies days_to_death=NA
Reduced copy number sample count by 824, to correct erroneous counting of tangent-normalized SNP6 data (will be in next run)
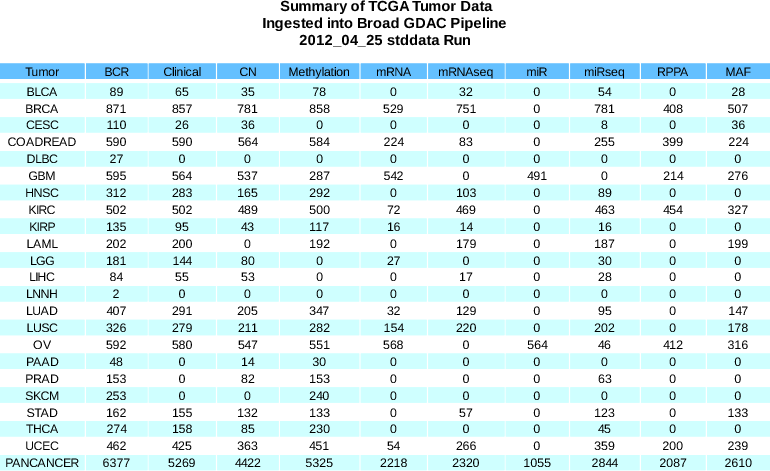
- 128 new methylation samples, including fix for missing 450 platform Beta values
- 824 new copy number samples
- Initial bundling of RPPA data type, with 2087 samples packaged for proteomics QC review
- Phase 1 of companion SDRFs implementation: phase 2 in next data run will complete implementation.
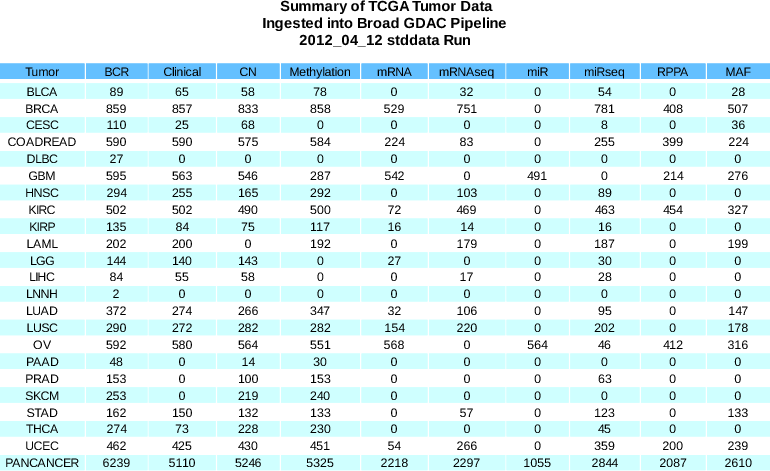
- Almost 800 new methylation samples: Human Methylation 450 platform included for first time
- MAFs for stomach (STAD, 133 samples) and cervical (CESC, 36 samples) included for first time
- New firehose_get utility, for simpler downloads.
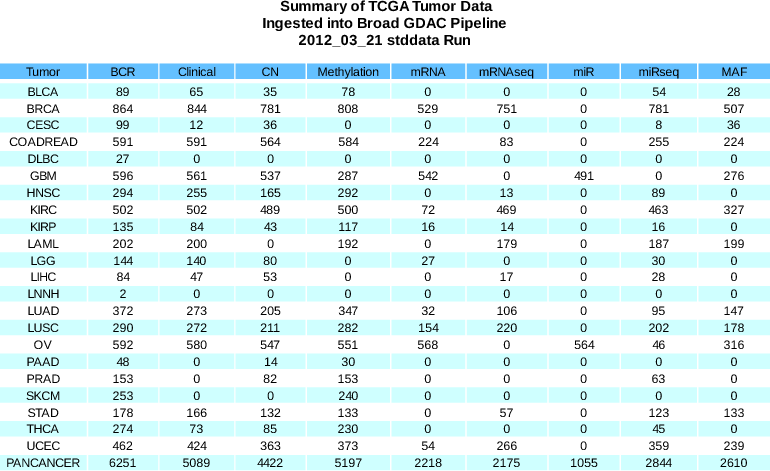
- Draft PANCANCER dataset included: 34 of 40 possible pipelines ran to completion, for an 85% success rate. At present this represents a simple aggregation of all tumor data, e.g. irrespective of hg18 versus hg19 alignment, but we will be advocating in followup TCGA discussions that these and other distinctions (like platform) will need to be considered more carefully in order to maximize scientific accuracy.
1100 more methylation samples
First samples of bladder cancer (BLCA) mutation data: 28 in all
To simplify access, all public archives from our stddata runs are now also hosted directly from Broad servers; no Level_1, Level_2, or Clinical PHI data is included, and credentialed download from the DCC remains the only way these protected data may be obtained.
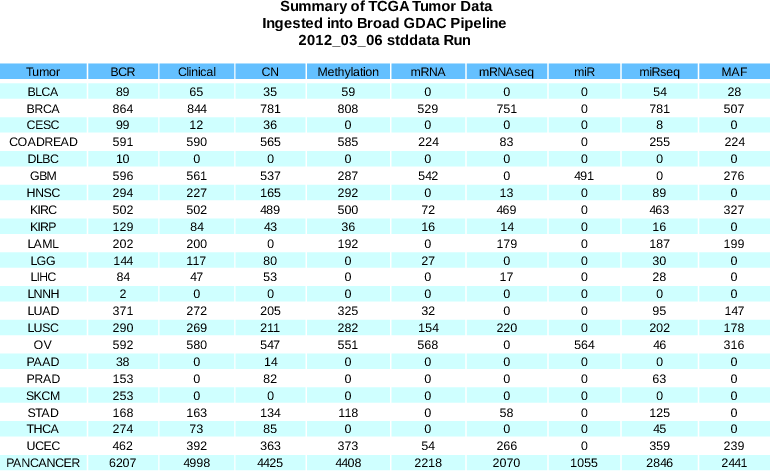
- Due to low sample differentials in early Feb we performed only 1 stddata run this month.
Methylation sample count increased by more than 1000 to 3290.
mRNAseq sample count increased by more than 300 to 2070.
Approximately 300 new BCR and clinical data samples.
With clinical data elements remaining stable since Sept 2011 for many tumor types, we've resumed execution of our Tier1 CDEs picker pipeline, with this including CDEs for 14 tumor types (versus only 8 tumor types when Tier1 CDEs were last packaged in summer of 2011)
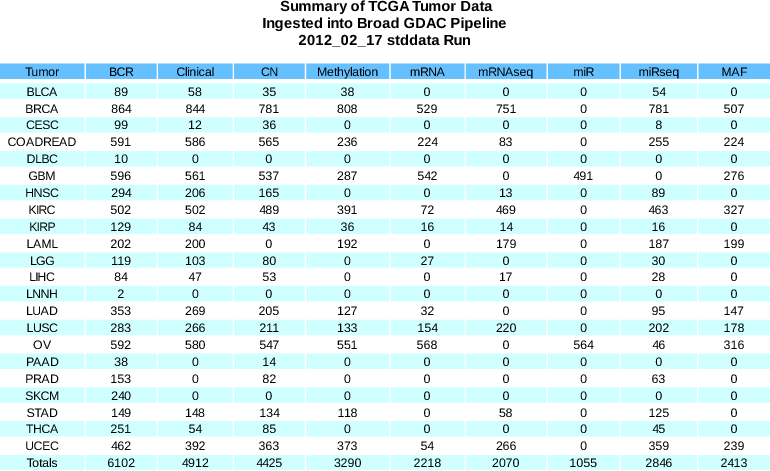
Absolute counts for Jan 2012 data runs have changed very little from December 2011.
As of 2012_01_24 run we believe redactions & rescissions are accurate to ~100% of DCC annotations database (with thanks to M. Ferguson for assistance). Please report to gdac@broadinstitute.org if you find otherwise.
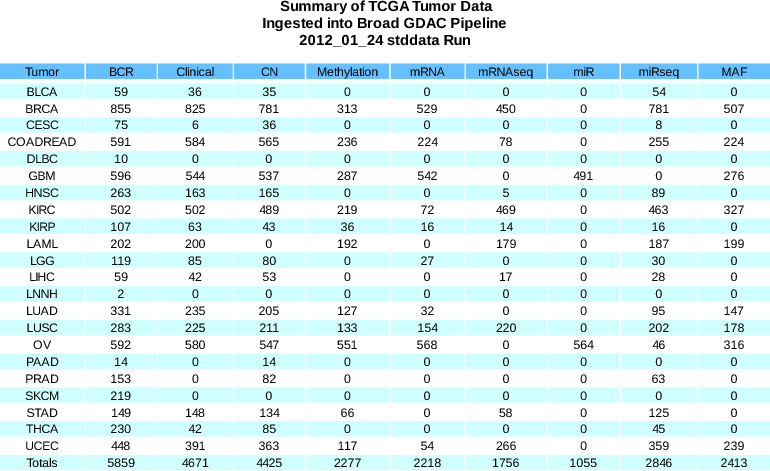
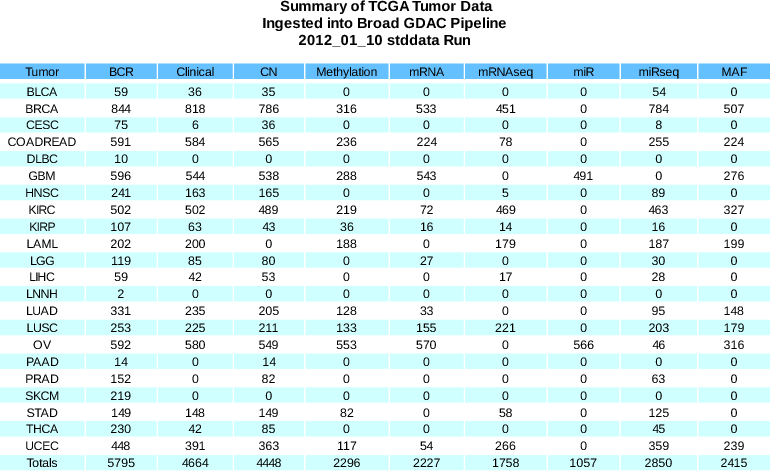
- To simplify data retrieval, the Tumor Type cells in our dashboard now link directly to the DCC locations from which the corresponding archives may be downloaded.
- All MAF and WIG files that are available to and processed by the Broad GDAC are now bundled within our stddata package.
In the 2011_12_30 data run the LUAD MAF count dropped to 148, as we corrected an error that resulted in the elimination of 110 spurious LUAD mutation samples.
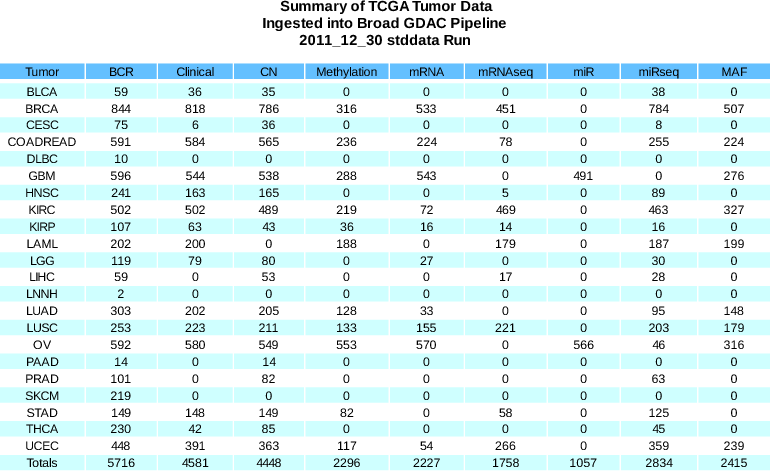
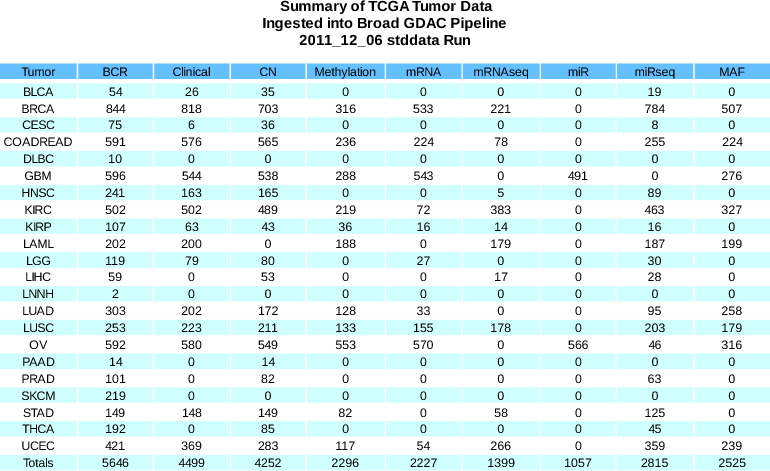
Due to ongoing software development and manual data collection efforts, no RNA-Seq or MAF data was included in the Nov 15, 2011 stddata runs.
RNA-Seq data were bundled in the Nov 28 stddata run, and both RNA-Seq and MAF data will be bundled with the December 2011 runs.
Clinical data have been bundled as of Nov 28 as well.