Release notes for the standardized data upon which these analysis runs are based are available here
| Anchor | ||||
|---|---|---|---|---|
|
| Panel | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| |||||||||
NOTE: The LIHC RPPA data submitted by MDACC early in 2016 were discovered to be mislabelled MESO samples. Thus the 2016_01_28 analyses and standard data pipelines for LIHC have been re-run using the corrected samples submitted in March, and the nozzle reports now contain notices of the discrepancies. The following table shows the changes in sample counts for RPPA data as a result of this patch:
|
| Anchor | ||||
|---|---|---|---|---|
|
| Panel | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||
|
...
| Panel | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||
|
...
| Panel | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
|
...
| Panel | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||
|
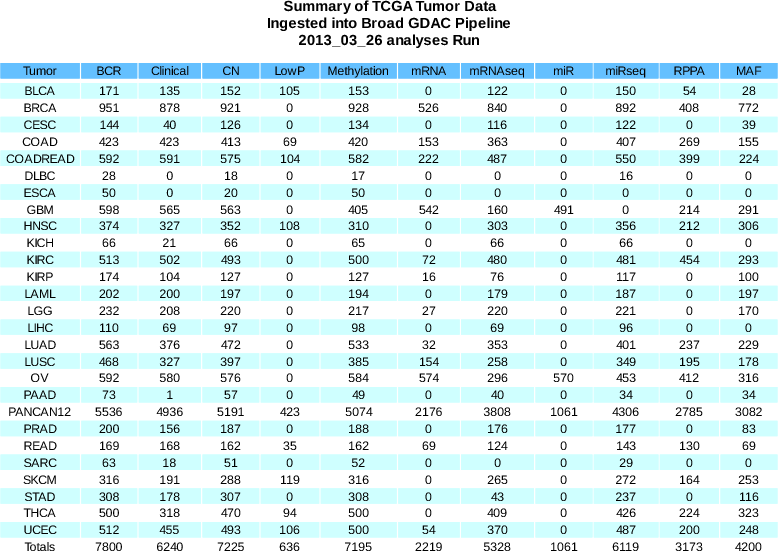
| Anchor | ||||
|---|---|---|---|---|
|
| Panel | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
|
...
| Panel | ||
|---|---|---|
| ||
|
| Anchor | ||||
|---|---|---|---|---|
|
| Panel | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||
|
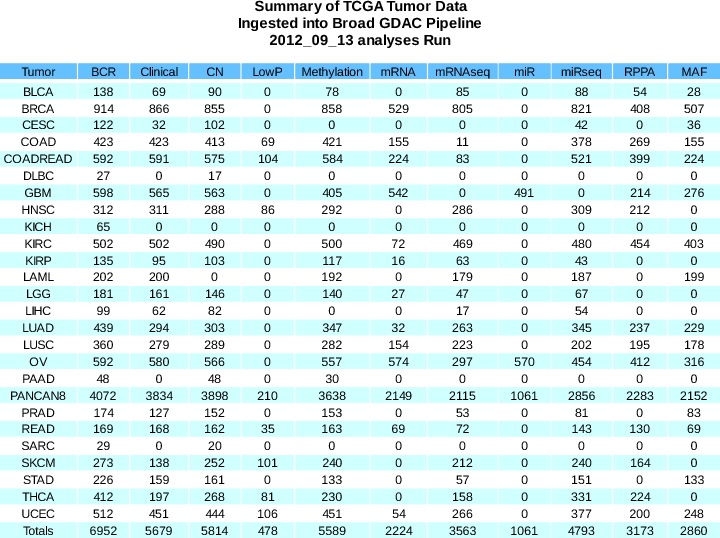
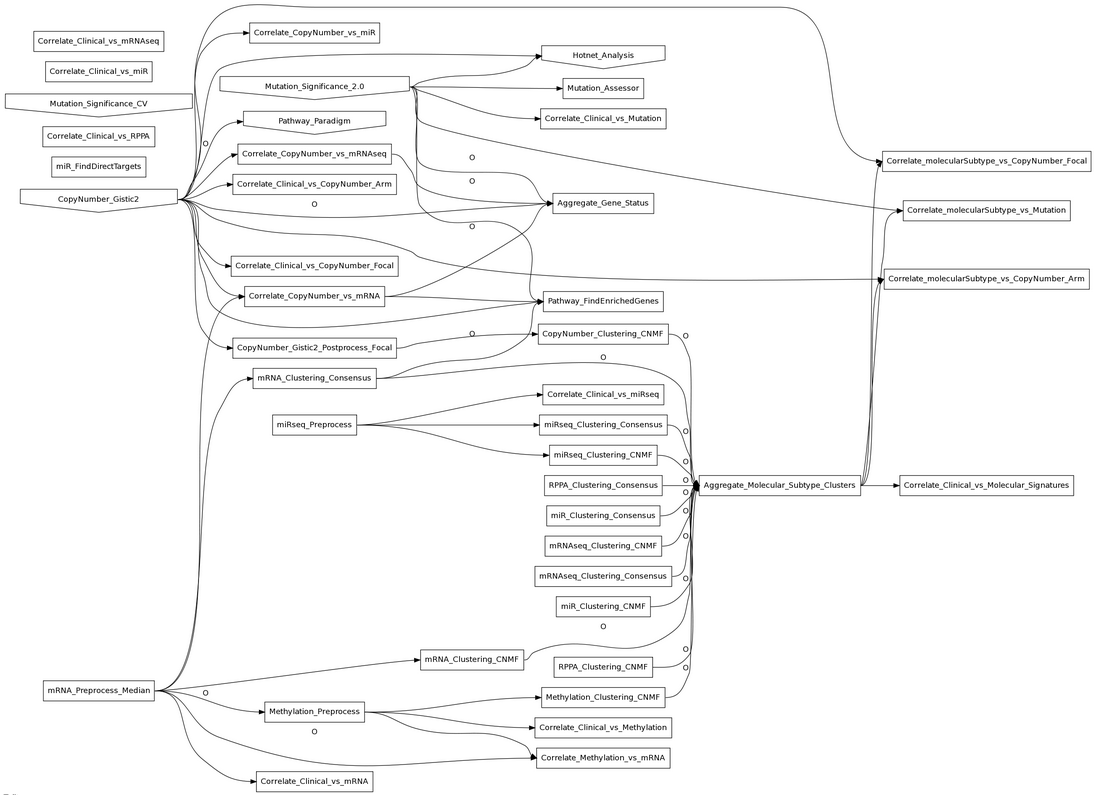{kind=link}
...
| Panel | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||
|
| Anchor | ||||
|---|---|---|---|---|
|
| Panel | ||
|---|---|---|
| ||
|
...