Frequently Asked Questions
...
| Panel |
|---|
Q: Where can I find a copy of the blacklist used in MutSig runs?A: The blacklist is used to filter out recurrent mutation sites that the MutSig development team found to cause issues with the determination of significance. Because these by nature include germline mutations that may not have been part of available databases at the original generation of the MAF, we are not permitted to release it to the public. |
| Panel |
|---|
Q: Why does your table of ingested data show that disease type XYZ has N methylation samples?A: We ingest and support both of the major methylation platforms (Infinium HumanMethylation450 and HumanMethylation27), therefore the entries in our data table give the sum of both. However, as noted in our June 2012 release notes, Firehose does not yet include the statistical algorithms used by TCGA AWGs to merge both of these methylation platforms into a single bolus; until those are shared we prefer |
| Panel | ||||||
|---|---|---|---|---|---|---|
A: Since inception Firehose analyses have been executed upon tumor samples and then correlated with clinical data. Nearly all analyses utilize primary solid tumor samples (numeric code 01, short letter code "TP" as given in the TCGA sample type codes table), with two exceptions:
Also note that each stddata run dashboard contains a samples summary report, which explains why – even though our GDAC mirrors ALL data from the DCC on a daily basis – not every sample is ingested into Firehose*. *Specifically, we filter out ALL samples listed as Redacted in the TCGA Annotations Manager, and FFPE samples are only available in standard data archives, not analyses. Programmatically, the FireBrowse patients api will give you a list of all patients in each cohort, either in bulk (all cohorts) or any subset you chose. It doesn’t give the complete aliquot barcode yet, but will in the very near future. In addition to playing with this API interactively through the FireBrowse UI, there are also Python, Unix, and R bindings, and even a pip-installable package. |
| Panel | ||||||
|---|---|---|---|---|---|---|
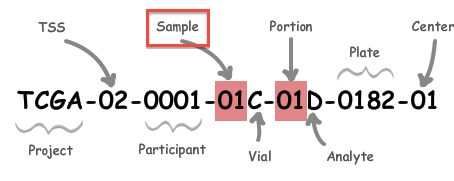
A: All analysis-ready patient samples are available in our stddata archives; control samples are not. You can obtain the stddata archives using our firehose_get utility or by traversing the FireBrowse user interface or stddata API. Each sample in the archive is identified by a TCGA Barcode that contains the sample type. As shown below, the Sample portion of the barcode can be looked up in the sample type code table available here (as can the tissue source site, aka TSS, et cetera). In addition, FireBrowse makes much of this information available programmatically in its metadata API. TCGA Barcode Description: As described here, a batch is uniquely determined by the first shipment of a group of analytes (or plate) from the Biospecimen Core Resource. So, in most cases the plate number of a sample is effectively synonymous with the batch id of the sample; an exception to this is when additional analytes from a participant are subsequently shipped the batch id will remain fixed at the first plate number.
|
| Panel |
|---|
Q: Where can I find additional information about a TCGA sample/analyteA: While the TCGA was active samples notes (aka annotations) had been maintained at the TCGA Annotations Manager. In July of 2016, however, the data portal of TCGA went offline, with all data (including annotations) having been migrated to the Genomic Data Commons. Please contact the GDC staff for details on how annotation information may be obtained after July 2016. |
...
| Panel |
|---|
Q: How are the copy number data generated, and what do their file names mean?A: This is discussed in the application note posted here: http://www.broadinstitute.org/cancer/cga/copynumber_pipeline. Note that the 'minus_germline, or 'nocnv' segment files, refer to whether the steps in section 2.3 are applied. The steps in section 2.4 are applied regardless. |
| Panel |
|---|
Q: What centers are responsible for sequencing XYZ tumor?A: Internally at the Broad we maintain /wiki/spaces/CGW/overview#CancerGenomeWorkgroup-Tumor_Sequencing_Centers If you are outside the Broad please consult the TCGA site for more information. |
...
| Panel | ||||||
|---|---|---|---|---|---|---|
A: RPKM and RSEM are different methods for estimating expression levels from mRNASeq data. RPKM (Reads Per Kilobase per Million mapped reads) is described in a paper by Mortazavi, Williams, McCue, Schaeffer & Wold titled Mapping and quantifying mammalian transcriptomes by RNA-Seq. RSEM (RNA-Seq by Expectation-Maximization) is considered by many to be a better estimation method and, if available, RSEM data is preferentially used in our downstream analyses. It is described in a paper by Bo Li & Colin Dewey titled RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. |
| Panel |
|---|
Q: How do I map mRNA isoform IDs to genes?A: We do not provide a mapping table for this, because in TCGA these data are generated by the University of North Carolina. However, for internal analyses we and others frequently use the UCSC Table Browser, e.g. as described in this BioStars recipe. |
...