1.1.40 (2019_10_13)- JavaScript Library Dependency Upgrades:
- jQuery 1.10.2 → 3.1.1
- jQueryUI 1.10.3 → 1.12.1
- fancyBox 2.15 → 2.17
- Removed fancyBox 2.15 from the repo, and added 2.17 as a submodule
- pip version set to 18.1
- earlier and later versions have issues with pylint 1.5.1
- Bugfixes:
- By sharing a common buttonset, the bulk-download modal broke the buttons on the archive-download modal in multiple ways if it was instantiated. Separate container classes were created for the buttons, and the buttons for each were given unique names and ids to prevent the previous collisions. New functions were written to simplify this process.
- A function that was only meant to apply to the reports-accordion was being applied to all context menu right-clicks and throwing errors in the console. It was updated to be more specific.
1.1.39 (2018_12_18)- Updated deployment code to be more user-friendly.
1.1.38 (2018_09_07)- Deployed iCoMut_Beta-0.21, correcting typos in panes
1.1.37 (2017_12_14)- Deployed iCoMut_Beta-0.20, to address rendering of legends in newer Chrome and Opera browsers
Code generator is now runnable/installable as a standalone tool (named fbgen); and has been verified to work for GTEX portalClient bindings version discernable via new client_version() endpoint, in both fbget cli tool and firebrowse module; this supplements the longstanding version info embedded within -v CLI option and heartbeat() endpoint - High level endpoints can now be invoked at CLI with 'help' to obtain same help that has historically been available by invoking endpoints with zero arguments
1.1.36 (2017_08_21)Added OLD RUNS link to top-level menu, so users can quickly contrast legacy run results with what FireBrowse currently displays
1.1.35 (2017_08_16)- Released v0.1.9 of fbget Python and Unix bindings to reflect client-side changes for WinXX and Mac/OSX
1.1.35 (2016_09_27)- In light of TCGA data portal shutdown, added new metadata/TSSites function to identify tissue source sites
- Released v0.1.8 of fbget Python and Unix bindings to reflect this new API (both zipfile and PyPi package)
1.1.34 (2016_08_12)- Deployed several new iCoMut instances in support of AWG analyses (TCGA credentials required to view)
- Boosted performance of some copy number API queries by creating additional indexes on the database backend
- Added to top level page a link to the detailed samples report generated during pipeline data ingestion.
1.1.33 (2016_06_13)- Avoid a race condition when redirecting to FireBrowse home page with
&download_dialog=true
- Enhanced the command line help mechanism to (a) be more robust and (b) allow more than 1 function to be queried at a time
1.1.32 (2016_06_02)- Minor internal patch, locking version of Google charts api to v42, to maintain stable featureset of sample counts plot.
1.1.31 (2016_05_18)- Minor internal patch to re-establish portability to older JavaScript v5 clients
1.1.30 (2016_05_13)- Major updates to iCoMut:
- integrated MutationSignature panel, to show nucleotide changes
 - support JSON input
- and more flexible JSON-based configuration, to further simplify deployment on other sites
- simpler visual customization, with new controllers (icons) along left edge of each panel, including:
- toggle (show or hide) samples which have zero mutations
- toggle display of outliers in mutation rate, so that hypermutated samples do not dominate
- more crisp, responsive and predictable drag/drop of panels
- cleaner and more readable popup windows when hovering over cells
- legends added for single-row panels (clinical variables, clustering results)
- generally faster rendering
- Updates to backend database and APIs:
- MutsigCV calls dropped from database:
- FireBrowse now serves only MutSig2 and MutSig2CV values
- But MutSig1.5 and MutSigCV results are still available as archives, e.g. retrievable with firehose_get
- Improvements to the mutation samples (MAF) api:
- Significantly better performance: previously could timeout on gene-level queries
- But now returns in fractions of a second
- Also changed the layout of returned data, to reflect the ordering of a TCGA compliant MAF. By default only a subset of the compliant MAF columns are returned, reflecting those of most common use.
- A different set of columns can be retrieved by specifying their individual names in the
column parameter, or all to retrieve everything. - The complete list of MAF columns available through is given in the new Metadata/MAFColNames method.
- The significantly mutated genes (SMG) api was made more flexible:
- previously, the
cohort parameter was unconditionally required and gene was completely optional - but now they are both semi-optional: for the call to succeed at least one of
cohort or gene must be supplied - this makes it very easy to compare, with a single call, how the mutation of a given gene varies across all cohorts
- Removed the Metadata/Cohort API; what it returned was a subset of the Metadata/Cohorts API
- The
gene parameter is now mandatory for the Samples/mRNASeq API: calling with just a barcode is no longer supported - Likewise, the
mir parameter is now mandatory for the Samples/miRSeq API
- Python/UNIX client bindings:
- Increased value of
page_size parameter used internally from the API default of 250 records per page to 1000. The original value of 250 yields a more responsive and robust experience in browsers, but is needlessly low for the programmatic use case where throughput of completing the entire query is a more important than conserving size. - Expose page_size parameter as top-level parameter to fbget: can then be used for any other API call without needing to specify
- Released version 0.1.6 to PyPi, reflecting these and the RESTful API changes described above
A series of small updates to the online tutorial - Top level UI now links to results for TCGA analysis working groups (AWGs). TCGA credentials are still needed to view unpublished results.
1.1.28 (2016_04_22)- Ingest Spring 2016 Firehose analysis run results
- iCoMut:
- loaded 4 additional disease cohorts: DLBC, ESCA, SARC, and THYM
- Completed most of work for major new release, stay tuned for announcement next week, incorporating many graphical and data exploration enhancements
1.1.27 (2016_03_31)- Added sort parameter to sample Counts api, defaulted to 'cohort' to ensure that samples plot on landing page stays in sorted order.
1.1.26 (2016_03_30)- Data download dialogs now support SendTo option to export data to GenomeSpace: for example
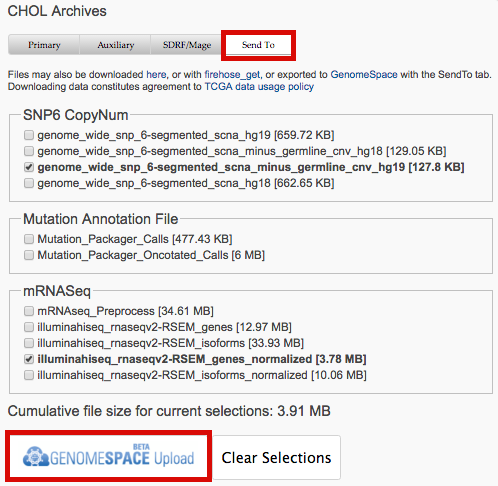 - The online tutorial was updated to reflect this, as well as new-ish advanced search features.
- Restored alphabetical-sort order to cohorts on samples plot of landing page.
1.1.24 (2016_03_02)- Loaded Firehose 2016_01_28 stddata run
- Numerous internal updates to further simplify deployment, which e.g. will help provision AWG-specific databases
- The HeartBeat API function now includes: the directory from and time at which the API server was launched
- Dropped beta clause from version
1.1.21 beta patch (2015_12_21)- No changes to FireBrowse codebase, but back-end mRNASeq data for MESO and SKCM were patched (per the 2015_11_01 patch release notes)
1.1.21 beta (2015_12_01)- Loaded Firehose 2015_11_01 stddata run
1.1.20 beta (2015_11_20)Loaded Firehose 2015_08_21 analysis run - iCoMut: several important new features, including:
- Significantly more advanced search: including logical inclusion/exclusion, and searching individual panel results using comparison operators (e.g. >, =, etc)
- Figure generation (download SVG), with embeddable link one can use to fully reproduce the figure
- Ability to insert additional genes into the mutation panel (as long as they are in the MutSig SMG list)
- Ability to display continuous data in addition to categorical (e.g. clinical age values are no longer just "hi/med/low" but actual numeric values)
- viewGene: several bug fixes and improvements to cross-browser support
Small cleanup in analysis report accordion of UI: remove duplicate Pathway_Overlaps report, and shrink MutSig report name entries Analyses/MAF api no longer shows results from MutSig_1.5 Analyses/FeatureTable and Analyses/Reports apis are now sorted in descending date order Metadata/Platforms and Metadata/Centers apis are now sorted, too, for easier visual inspection
1.1.19 beta (2015_11_12)- Address viewGene bug, where filter selector was not rendering in safari and firefox
1.1.18 beta (2015_11_12)- Fix viewGene bug, which would cause it to hang while rendering expression plot
1.1.17 beta (2015_09_27)- Ingest the August 2015 Firehose standard data run:
API through which Firehose-picked and normalized clinical data are accessible has been renamed to Samples/Clinical_FH Verbatim TCGA clinical data may be accessed through the new Samples/Clincal API A CDE will be reflected in either API only when it has a value other than NA for at least 1 patient case in any disease cohort. - For backward compatibility, the Samples/ClinicalTier1 remains available (as a synonym for Samples/Clinical_FH)
- fbget Python and UNIX CLI bindings have been suitably updated
- Which makes it extremely easy to determine for what patients and cohorts any given CDE is defined: e.g.
fbget clinical cde=gleason_score
will show each patient case with a valid gleason_score, across all cohorts
- viewGene: now enforces rendering of at most 1 gene per Submit; if multiple genes are given the first is selected
- iCoMut:
- Popup tooltips for mutation panels now show total # of mutations AND fractional % of a given type (e.g. missense)
- New search feature, enabling one to see into which clusters/panels/etc a given patient (or set of patients) falls

- After extensive testing, upgraded backend database from Mongo2.x to Mongo3.x: v3 dramatically reduces the memory footprint and data storage sizes, which yields greater performance and also clears considerable breathing room to add more data APIs (e.g. for methylation, RPPA, etc) as well as AWG-specific databases
- Fixed bug GDAC-3204: counts api was returning empty results when both
sample_type= and cohort= parameters were specified
1.1.15 beta (2015_08_24)- Added Code, CenterType, and ShortName fields to the payload returned by the Metadata/Centers api function
- The HeartBeat function now returns the version number and build time/date, too
- Re-ingested data served by Clinical samples function, to filter non-ASCII characters
- To address this viewGene issue, insert gene name into metadata to ensure cache is suitably invalidated.
- Addressed a performance issue that had crept into the FireBrowse landing page: the sample counts barchart was noticeably slow to load, apparently since mid-August due to a typo in the font name. Strangely, the typo has been there all along, but the landing page has only been slow for about 1 week. It is fast again.
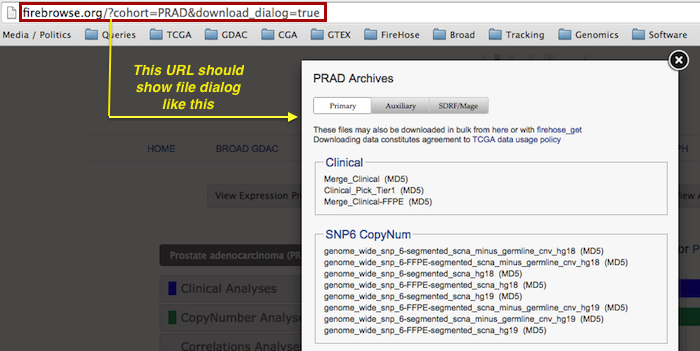
1.1.14 beta (2015_07_23)- Fixed the
download_dialog feature of the landing page, so that data browsing redirects from the GDAC homepage once again pop up dialogs resembling this:
- Updated the Clinical samples API to reference the new interactive CDE table, which on a single page shows exactly what CDEs are available for all disease cohorts.
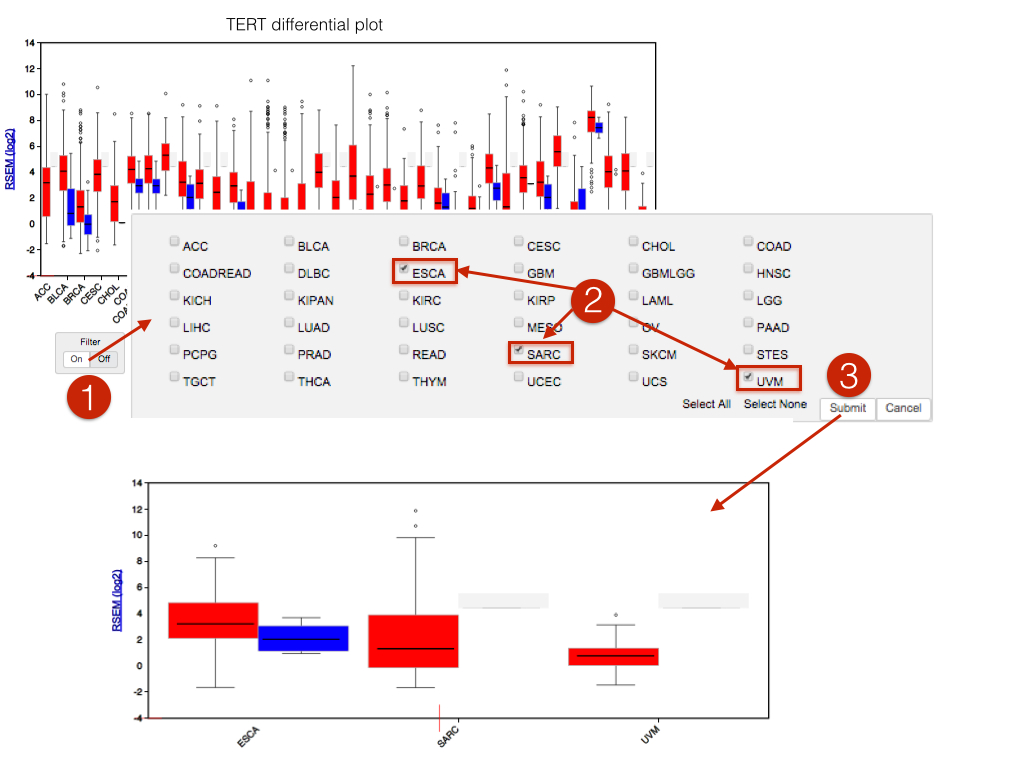
1.1.13 beta (2015_07_10)viewGene now has a cohort filter control, allowing subsets of cohorts to be viewed:
 iCoMut now properly shows LAML data; and includes a few other small fixes - Updated the online tutorial.
1.1.12 beta (2015_06_29)Enable password protection when displaying (unpublished) analysis working group (AWG) results
1.1.11 beta (2015_06_24)- Updated to reflect June 2015 stddata run.
- fbget Python and UNIX cli bindings updated to v0.1.4, and now also installable from PyPi via the pip tool.
1.1.10 beta (2015_06_09)- New control added to viewGene, enabling it to visualize RPKM format expression data; as some cohorts (e.g. ESCA, STAD, STES) do not have RSEM expression data.
1.1.9 beta (2015_06_04)- Internal modifications to simplify deployment
1.1.8 beta (2015_06_01)- Remove scale control from viewGene widget
1.1.7 beta (2015_05_18)- Make rawMAF datatype visible in user interface and API
1.1.6 beta (2015_05_15)- FeatureTable api: fixed GDAC-2988 (selecting columns was causing exception, reported by M. Deng)
- SMG api : changed the default to sort by rank instead of q value
- Initiated this release notes page, visible from the FireBrowse home
1.1.5 beta (2015_05_11)Incorporated Spring 2015 Firehose Analysis run: over 8000 additional aliquots analyzed; added 7 new analysis tasks and over 300 analysis reports (nearly 1500 total) - iCoMut: a powerful new synoptic tool for interpretation and exploration CoMut figures have quickly become a staple of TCGA research. Within a single graphic they provide a comprehensive analysis profile, enabling the reader to quickly infer relationships between co-occurring results. With
iCoMut researchers can now explore coMut figures interactively, sorting and reordering samples and results as they see fit. - New viewGene expression visualizer: built on top of the FireBrowse RESTful API,
viewGene generates a boxplot of mRNASeq expression levels for a selected gene across all cohorts. The fbget suite of automatically-generated, open source wrappers to the FireBrowse RESTful API. fbget makes it even easier to ask both coarse- and fine-grained questions about TCGA data and Firehose analyses, from Python and the UNIX command line. - Reports and FeatureTable apis now make older results available, too, not merely those from the latest analysis run. The search criterion is YYYY_MM_DD datestamp.
- We are also very happy to mention FireBrowseR, a set of open source R bindings to FireBrowse developed by a computer science Ph.D. student in Germany!
1.1.4 beta (2015_04_23)- Loaded with Spring 2015 Firehose StandardData Run run
- Rationalized "requiredness" to be consistent across entire API:
- Now, nearly all functions require that at least one gene OR cohort OR barcode be specified
- This increases query performance, and clarifies API semantics
- New Quartiles API for mRNASeq expression, providing a summary of expression across all samples in a disease cohort, suitable e.g. for boxplot visualization
- Corrected docs for SampleTypeBarcode function to indicate that it takes only 1 barcode (as a "path parameter")
- Changes to the default sort parameter for several API functions:
- mRNASeq api and miRSeq functions now default to disease cohort instead of gene and mir, respectively
- significantly mutated genes and mutation file functions now default to disease cohort instead of Q value and gene, respectively
- When iterating over multiple pages of TSV, CSV output, the api will now return a header for ONLY the first page
- Columns containing commas are now delimited with double quotes in CSV return payloads
- Increased tolerance for messy input: distinct values of multi-valued parameters (e.g. gene=egfr,tp53,BRCA1 in mRNASeq samples api) may now be delimited by arbitrary whitespace (in addition to commas, as previously supported)
- In the interactive docs UI the page and page_size parameters are no longer required by any API function: they continue to default to reasonable values for the common case.
- The payload of the SampleTypes api now returns the description of the sample type (e.g. TP = tumor primary)
An empty response is now returned when no records in FireBrowse match a given query; previously a payload such as
would be returned in such cases (e.g. for this mRNASeq query). |